Introduction
Rhodospirillum is a gram negative bacteria of the genus of photosynthetic bacteria of the family Rhodospirillaceae. Their cells are generally spiral-shaped, polarly flagellated and contain vesicular, lamellar of stacked photosynthetic membrane. One of the type species of this genus is Rhodospirillum rubrum (Rsp.) rubrum, an anoxygenic phototrophic purple bacterium with a long history as a model for the study of bacterial photosynthesis and related metabolic processes. It is unique among purple bacteria by producing both rhodoquinone (RQ) and (UQ) as electron carriers and bacteriochlorophyll (BChl) a esterified at the propionic acid side chain by geranylgeraniol (abbreviated as BChlaG) rather than phytol.[1]
This organism contains chlorophyll b, which is different than chlorophyll a found in plants. Chlorophyll b distinguished by a lower absorption spectra, absorbs maximally at 660 nm rather than at 680 nm. Anoxygenic phototrophs such as R. rubrum can contain several bacteriochlorophylls, and most purple bacteria have bacteriochlorophyll a, which absorbs maximally between 800 and 925 nm. Organisms with many different types of chlorophylls are at an advantage, because they can use more of the energy of the electromagnetic spectrum. [2]
Function
The light-harvesting complexes (LHC) of photosynthetic purple sulfur and non-sulfur bacteria are responsible for the highly efficient collection and transfer of light energy to the photosynthetic reaction centres. This results in an initial separation of charge in the reaction centre (RC) and ultimately conversion of the light energy into a chemically useful form [3].
Rsp. rubrum has a single pair of αβ-polypeptides in its core light-harvesting (LH1) complex and a reaction center (RC) cytochrome (Cyt) c subunit present in many purple bacteria; thus, Rsp. rubrum is one of the simplest phototrophic bacteria known, in terms of its photosynthetic light reactions. Because the entire Rsp. rubrum LH1 complex and a stable B820 LH1-subunit can be reconstituted using the αβ-polypeptides and pigment molecules. Both complexes have been intensively studied as models of the bacterial antenna apparatus and as such have provided a wealth of information on mechanisms of light energy acquisition, pigment−protein interactions and assembly of multicomponent complexes. [4]
The minimum photosynthetic unit in these bacteria is composed of a reaction center (RC) surrounded by a light-harvesting complex called LHC1. The two are commonly considered in the scientific literature as a unit, which together form the so-called RC-LH1 complex (in proportion 1:1). This is capable of converting light energy in partnership with a second electron transfer protein embedded in the membrane, the so-called cytochrome bc1 complex. The LH1 complex Bchls absorb light around 875 nm.
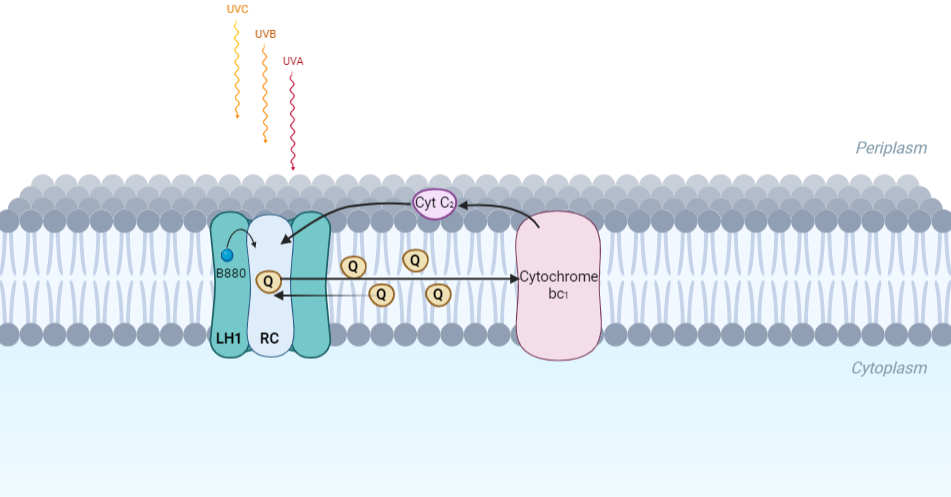
Representation of the cyclic electronic transport process in the intracytoplasmic membrane of purple bacteria. Light is absorbed by the LHC1 complex, which transfer their excitation energy to the reaction center, where a separation of loads. The light energy absorbed by carotenoids and bacteriochlorophylls (B880) generates a change in the energy state of the molecules that can be transferred following several excitation pathways between the photosystem pigments until it ends up reducing the ubiquinones located in the RC (thus converting light energy into chemical energy). They escape through interprotein pores in the RC to transfer electrons to Cyt b1. The route is completed by a soluble protein (Cyt c2) that ends up donating electrons to LH1.
Inicial Structures
For a long time, structures of both purified LH1 and the RC-associated core complex (LH1-RC) of Rsp. rubrum have not been obtained at high resolution, and no RC atomic structure was known. The 8.5 Å resolution projection of R. rubrum LHCl represents the first glimpse of the structural architecture of the fundamental building block of the photosynthetic membrane in purple BChla-containing bacteria. The crystals diffract beyond 8 Å and the projection map was calculated to 8.5 Å. The projection map shows 16 subunits in a 116 Å diameter ring with a 68 Å hole in the center.
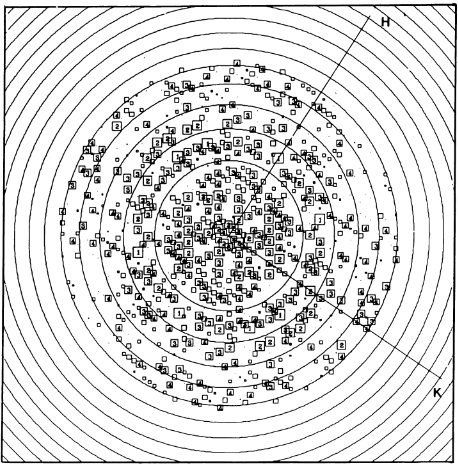
Fourier components measure up to 8.0 Å resolution for estimation. The size of the boxes indicates the [IQ][[1]] value, with IQ=1 being the largest. This image shows the equivalent projection calculated from the average amplitudes and phases of eight images with symmetry applied and amplitudes enhanced by a resolution-dependent scaling factor. The positive density rings can represent the protein. 16-fold strong and 32-fold weak components can be seen above the noise level, thus indicating a 16-fold non-crystalline rotational symmetry in projection density. The 16 times rotationally filtered map is shown below.
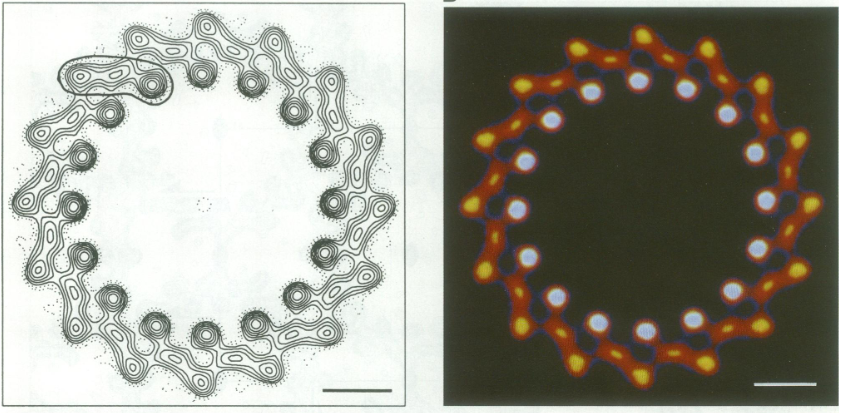
Each of the 16 subunits contains three distinct domains. Assuming that the ring in our projection represents a 3 Da cylinder with outside diameter 116 Å, inside diameter 68 Å and length 45 Å (one membrane layer thick), this would correspond to a volume of 311000 Å3. Assuming an average protein density of 0.77 Da/A3, this would give the cylinder a total mass of 266 kDa, corresponding to ~17 αβ-subunits with their associated bacteriochlorophylls and carotenoids, in reasonable agreement with our observation of 16 subunits [5].
Cryo-EM
Cryogenic electron microscopy (cryo-EM) is a cryomicroscopy technique applied to samples cooled to cryogenic temperatures. For biological samples, structure is preserved by embedding in a glassy ice environment. An aqueous sample is applied to a mesh grid and frozen by immersion in liquid ethane or a mixture of liquid ethane and propane [6]. This technique has advanced dramatically to become a viable tool for high-resolution structural biology research. The ultimate outcome of a cryo-EM study is an atomic model of a macromolecule or its complex with interacting partners. Recent advances in direct electron detectors as well as reconstruction single particle algorithms have led to the determination of the structure of macromolecular complexes ranging from 2 to 5 Å resolution. At these resolutions, also known as “near atomic” resolution, it is possible to infer all-atom structures de novo.
The first step in cryo-EM structure determination is de novo structure determination, where an initial model can be built, given only one sequence and a reconstruction, when no other limited structural information is known. In the second stage, the model is optimized, where a wide range of class of methods for improving the fit of a model to the data and improving the geometry of a model. Finally, tools for model validation are described, in attempt to quantify the overall accuracy of a model given a reconstruction.
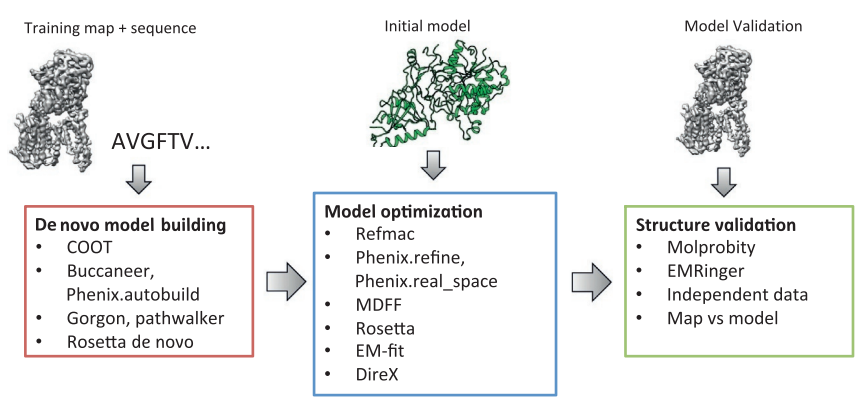
An overview of three steps of atomic model determination from near-atomic resolution data. (Left) De novo building methods take primary sequence and map, and automatically produce a backbone model with sequence registered, identifying which regions in the map correspond to particular sequences. (Center) Model optimization takes an initial model—either produced from de novo building, or from a highresolution homologue—and optimizes the coordinates to better agree with the map, as well as adopt more physically realistic geometry. (Right) Model validation aims to assess—both globally and locally—the accuracy of a model, given experimental data. Such tools are useful not only for assessing overall accuracy but also for tuning parameters of optimization [7].
Structure of Photosynthetic LH1-RC Super-complex of Rhodospirillum rubrum
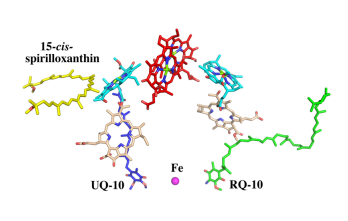
The cryo-EM structure of Rsp. rubrum LH1-RC was determined at 2.76 Å resolution. The LH1 complex forms a closed, slightly elliptical double ring composed of 16 pairs of α(inner)β(outer)-polypeptides, 32 BChls aG and 16 all-trans-spirilloxanthins, as we can se on the Fig. 1. The RC of Rsp. rubrum is surrounded by the LH1 complex with only a few close contacts on the periplasmic surface with residues near Ser34 in the LH1 α-polypeptides. There is no apparent strong interactions of Rsp. rubrum RC with your LH1 polypeptides (Figure 1 a-c) as occurs in Cyt c-bound LH1-RCs where the C-terminal domains of some LH1 α-polypeptides interact extensively with the Cyt c subunit. The BChl aG molecules in Rsp. rubrum LH1 form an elliptical, partially overlapping ring with average Mg−Mg distances of 9.3 Å within a dimer and 8.5 Å between dimers (Figure 1c). [8]
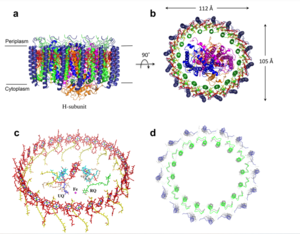
Fig. 1. Structure overview of the
Rsp. rubrum LH1-RC complex. (a) Side view of the LH1-RC parallel to the membrane plane. (b) Top view of the LH1-RC from the periplasmic side of the membrane. (c) Tilted view of the cofactor arrangement. (d) Superposition of Cα carbons of the LH1 αβpolypeptides between
Rsp. rubrum and (gray, PDB: 5Y5S). Color scheme: LH1-α, green; LH1-β, slate-blue; L-subunit, magenta; Msubunit, blue; BChl aG in LH1 and special pair, red sticks; Accessory BChl aG, cyan sticks; BPhe aG, light-pink sticks; Spirilloxanthin, yellow sticks; UQ10, blue sticks; RQ-10, green sticks; Fe, magenta ball. Phospholipids and detergents are omitted for clarity
For a more comprehensive view of the structure of Rsp. rubrum LH1-RC, this molecule can be visualized in different ways, such as, for instance, by the shape of , or .
The BChl aG molecules in Rsp. rubrum LH1 forms an elliptical, partially overlapping ring with average Mg−Mg distances of 9.3 Å within a dimer and 8.5 Å between dimers. These molecules are ligated by histidine residues (α-His29 and β-His39), as shown in the next image (Fig. 3). The geranylgeranyl side chains in the BChl aG associated with βpolypeptides form a tail-up conformation (Figure 2a). This may be due to the multiple double bonds present in the geranylgeranyl group, which results in a more rigid conformation. This unique conformation allows the geranylgeranyl side chains of the β-associated BChl aG to interact with the bacteriochlorin ring of the α-associated BChl aG (Figure 3b). This proximity likely allows for π−π interactions between the double bonds in the geranylgeranyl group and nearby bacteriochlorins. The C3-acetyl oxygen atoms in BChl aG form hydrogen bonds with the Trp residues (α-Trp40 and β-Trp48) of their associated polypeptides (Figure 2c, d), in agreement with the results of resonance Raman spectroscopy. The aromatic side chains of the Trp residues also interact with bacteriochlorins through π−π stacking, further stabilizing the LH1 complex. [9][10]
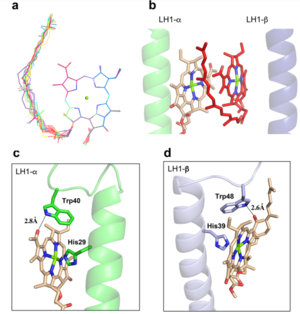
Fig. 2. (a) Superposition of the bacteriochlorin rings of 16 BChl aG molecules bound to the LH1 βpolypeptides. (b) Interactions between a geranylgeranyl side chain of the BChl aG (red sticks) bound to LH1 β-polypeptide and the bacteriochlorin ring (tint sticks) of a BChl aG bound to the LH1 α-polypeptide. (c) Coordination and hydrogen bonding of the BChl aG in LH1 α-polypeptide. (d) Coordination and hydrogen bonding of the BChl aG in LH1 β-polypeptide.
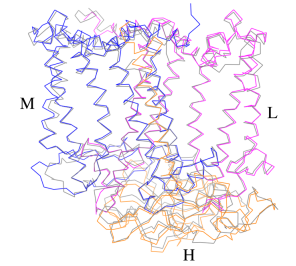
The Rsp. rubrum RC is composed of L, M, and H subunits with an overall protein structure and cofactor organization as reported in other purple bacteria.
A superposition of Cα carbons for the RC protein subunits between Rsp. rubrum (colored) and (gray, PDB: 3I4D), can be seen on the image below, and its cofactor arrangement on the next one. [11]
A total of 10 phospholipids (2 phosphatidylglycerols, 5 cardiolipins, and 3 phosphatidylethanolamines) were modeled in the cavities between the RC and LH1, within the range determined by biochemical analysis. Most cardiolipins were located on the cytoplasmic side of the membrane with their head groups pointing toward the membrane surface, whereas most phosphatidylglycerols and phosphatidylethanolamines were distributed on the periplasmic side. This phospholipid distribution is similar to that observed in other LH1-RCs. Structurally defined detergent DM molecules were identified in the density map, and most of them were located on the periplasmic side between the LH1 βpolypeptides with their head groups aligned on the presumed membrane surface. [12]
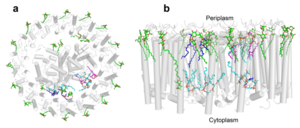
Fig. 3. Phospholipids, detergents, and channels in the LH1-RC complex. Top view (a) and side view (b) of the phospholipid and detergent distributions for CL (cyan), PG (magenta), PE (blue), and DDM (green). All proteins are shown in gray.
The Rsp. rubrum LH1 complex is particularly well-known for its ability to form a highly stable structural subunit with an absorption maximum of 820 nm. Two different subunit forms can be distinguished from the LH1 complex: a face-to-face and a back-to-back configuration for the bacteriochlorins. Solution NMR and reconstitution experiments established that the B820 subunit has the face-to-face configuration with π-overlap at pyrrole rings III and V.[13]
Due to its structural simplicity and flexibility, the Rsp. rubrum B820 subunit has been thoroughly investigated and the results from which predicted minimal requirements for stabilizing the subunit structure. These requirements included (i) a central α-helical transmembrane domain composed of 18 hydrophobic residues, (ii) a His residue for coordination and hydrogen bonding to different BChl molecules, (iii) a Trp residue for hydrogen bonding to the BChl C31 carbonyl oxygen, and (iv) N-terminal regions of α- and βpolypeptides. The His interaction was estimated to account for over half of the stabilization energy of the B820 subunit, followed by hydrogen bonding by Trp residues.[14][15]
Additionally, our Rsp. rubrum LH1 structure also underscores the importance of the C-terminal domains (Figure 4d), especially for the β-polypeptide where the amino acids are highly conserved. The side chain of β-Arg46 forms multiple hydrogen bonds with the hydroxyl group of α-Thr46 and main chain oxygen atoms of α-Arg37 and α-Ala44. β-Arg46 is conserved in the LH1 of almost all purple bacteria and contributes 2.0 kcal/mol stabilization energy to the B820 subunit and this follows only the pigment−protein interactions of BChl a/β-His39 (>6 kcal/mol) and BChl a/β-Trp48 (3.7 kcal/mol) in stabilization energy.[16][17]
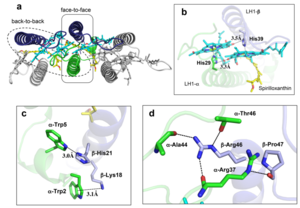
Fig. 4. (a) Two structural subunits with different configurations for the BChl aG dimers. Color codes in the subunits: LH1-α, green; LH1-β, slate-blue; BChl aG, cyan; spirilloxanthin, yellow. Other polypeptides and pigments are shown in gray. (b) Structure of the face-to-face subunit showing coordination and cross hydrogen bonding of the His residues to BChl aG molecules. (c) Major interactions in the Nterminal region within a face-to-face subunit. (d) Major interactions in the C-terminal region within a face-to-face subunit. Dashed lines indicate distances shorter than 3.5 Å.
References
- ↑ Singleton, P. and Sainsbury, D. (1987) Dictionary of Microbiology and Molecular Biology, second edition. John Wiley & Sons Inc., New York.
- ↑ Brock, T.D., Madigan, M.T., Martinko, J.M. and Parker, J. (2000) Biology of Microorganisms, ninth edition. Prentice Hall, New Jersey.
- ↑ 10.1002/j.1460-2075.1995.tb07041.x
- ↑ 10.1021/acs.biochem.1c00360
- ↑ 10.1002/j.1460-2075.1995.tb07041.x
- ↑ 10.1017/S1431927608080781
- ↑ 10.1016/bs.mie.2016.06.003
- ↑ 10.1021/acs.biochem.1c00360
- ↑ 10.1073/pnas.91.15.7124
- ↑ 10.1016/0005-2728(85)90048-9
- ↑ 10.1021/acs.biochem.1c00360
- ↑ 10.1016/j.bbabio.2019.04.001
- ↑ 10.1042/BCJ20160753
- ↑ 10.1021/bi9722709
- ↑ 10.1039/C7SC04905F
- ↑ 10.1023/A:1006337827672
- ↑ 10.1021/bi049798f











