This old version of Proteopedia is provided for student assignments while the new version is undergoing repairs. Content and edits done in this old version of Proteopedia after March 1, 2026 will eventually be lost when it is retired in about June of 2026.
Apply for new accounts at the new Proteopedia. Your logins will work in both the old and new versions.
Calculate structure
From Proteopedia
| Line 3: | Line 3: | ||
Any one page of Proteopedia can be run in the signed ver. 12 by appending "?JMOLJAR=http://chemapps.stolaf.edu/jmol/docs/examples-12/JmolAppletSigned0.jar" to the url of the page and reloading the page. The user must give permission for the signed version of Jmol to open, and when it does it has a red frank, whereas in the unsigned version it is grey. Click on the ''Jmol frank'', in the ''main menu'' which opens click on ''Console'', in the bottom box enter the commands: select protein; calculate structure; cartoon; color structure and then click ''Run''. | Any one page of Proteopedia can be run in the signed ver. 12 by appending "?JMOLJAR=http://chemapps.stolaf.edu/jmol/docs/examples-12/JmolAppletSigned0.jar" to the url of the page and reloading the page. The user must give permission for the signed version of Jmol to open, and when it does it has a red frank, whereas in the unsigned version it is grey. Click on the ''Jmol frank'', in the ''main menu'' which opens click on ''Console'', in the bottom box enter the commands: select protein; calculate structure; cartoon; color structure and then click ''Run''. | ||
</blockquote> | </blockquote> | ||
| - | '''Calculate structure''' is based on Defined Secondary Structure of Protein (DSSP), a program written in Pascal.<ref name=DSSP>W. Kabsch & C. Sanders, ''Biopolymers'', '''22''', 2577-2636, 1983.</ref> The secondary structure recognition algorithms are based mainly on hydrogen-bonding patterns along with geometric structures , such as bends. There are two different hydrogen-bonding patterns which are recognized. The one determines the value of n in the expression ''i'' + n (''i'' is a residue that forms a hydrogen bond with a residue n residues removed from residue ''i''.) where n = 3, 4 or 5. These values define three types of turns. A peptide segment that has repeating turns of the same type are called 3<sub>10</sub> helix, α-helix, or п-helix, respectively. If the turn is isolate, it is simply called an n-turn. The other recognized pattern is a hydrogen bond which is between residues which are not close together in sequence. This type of hydrogen bond is called a bridge. | + | '''Calculate structure''' is based on Defined Secondary Structure of Protein (DSSP), a program written in Pascal.<ref name=DSSP>W. Kabsch & C. Sanders, ''Biopolymers'', '''22''', 2577-2636, 1983.</ref> The secondary structure recognition algorithms are based mainly on hydrogen-bonding patterns along with geometric structures , such as bends. There are two different hydrogen-bonding patterns which are recognized. The one determines the value of n in the expression ''i'' + ''n'' (''i'' is a residue that forms a hydrogen bond with a residue n residues removed from residue ''i''.) where n = 3, 4 or 5. These values define three types of turns. A peptide segment that has repeating turns of the same type are called 3<sub>10</sub> helix, α-helix, or п-helix, respectively. If the turn is isolate, it is simply called an n-turn. The other recognized pattern is a hydrogen bond which is between residues which are not close together in sequence. This type of hydrogen bond is called a bridge. Kabsch & Sanders define a ladder as a "set of one or more consecutive bridges of identical type" and a sheet as a "set of one or more ladders connected by shared residues". |
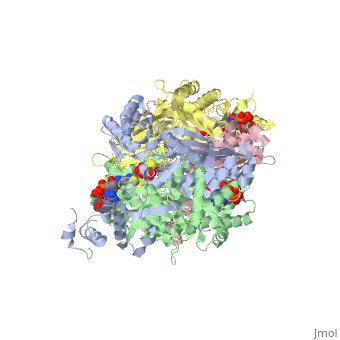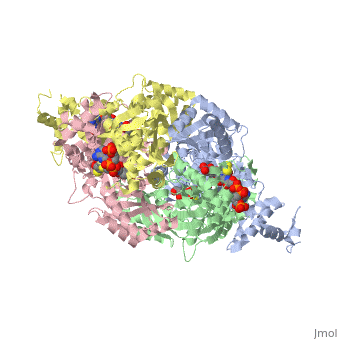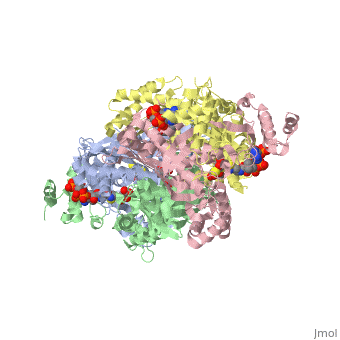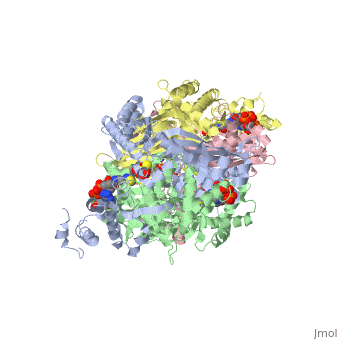
==Your Heading Here (maybe something like 'Structure')==<StructureSection load='1dq8' size='500' side='right' caption='Structure of HMG-CoA reductase (PDB entry [[1dq8]])' scene=''>Anything in this section will appear adjacent to the 3D structure and will be scrollable. | ==Your Heading Here (maybe something like 'Structure')==<StructureSection load='1dq8' size='500' side='right' caption='Structure of HMG-CoA reductase (PDB entry [[1dq8]])' scene=''>Anything in this section will appear adjacent to the 3D structure and will be scrollable. | ||
Revision as of 15:47, 30 June 2011
An important part of protein structure is the secondary structure which is made up of helices, sheets and turns and Jmol as described in Secondary structure is capable of determining and displaying these three types of structures. The Calculate structure[1] command which re-calculates the secondary structure does a more complete job of identifying and displaying the secodnary structures, is available in Jmol ver. 12, but is not available in Jmol 11.8 which is used in Proteopedia as of June 2011.
Any one page of Proteopedia can be run in the signed ver. 12 by appending "?JMOLJAR=http://chemapps.stolaf.edu/jmol/docs/examples-12/JmolAppletSigned0.jar" to the url of the page and reloading the page. The user must give permission for the signed version of Jmol to open, and when it does it has a red frank, whereas in the unsigned version it is grey. Click on the Jmol frank, in the main menu which opens click on Console, in the bottom box enter the commands: select protein; calculate structure; cartoon; color structure and then click Run.
Calculate structure is based on Defined Secondary Structure of Protein (DSSP), a program written in Pascal.[2] The secondary structure recognition algorithms are based mainly on hydrogen-bonding patterns along with geometric structures , such as bends. There are two different hydrogen-bonding patterns which are recognized. The one determines the value of n in the expression i + n (i is a residue that forms a hydrogen bond with a residue n residues removed from residue i.) where n = 3, 4 or 5. These values define three types of turns. A peptide segment that has repeating turns of the same type are called 310 helix, α-helix, or п-helix, respectively. If the turn is isolate, it is simply called an n-turn. The other recognized pattern is a hydrogen bond which is between residues which are not close together in sequence. This type of hydrogen bond is called a bridge. Kabsch & Sanders define a ladder as a "set of one or more consecutive bridges of identical type" and a sheet as a "set of one or more ladders connected by shared residues".
==Your Heading Here (maybe something like 'Structure')==
| |||||||||||