Putative kinase (3HDT) shows limited activity as a cytidylate kinase, utilizing ATP and dCMP as ligands.
Introduction
Kinases (or phosphotransferases) facilitate the transfer of a phosphate group from one molecule to another and are involved in cell growth and signaling. This work characterizes a protein (PDB ID 3HDT) with unknown function, and tests for kinase activity. The goal of this research is to characterize the protein, 3HDT, that has an unknown function. This research is part of the BASIL project that involved performing in silico and in vitro modules to make predictions and study the function of this protein.
In silico anaylsis
We used a variety of in silico tools with our protein, 3HDT to find similarities with other amino acid sequences, protein family matches, and structural comparisons to known proteins in the PDB. Below are the recorded results and information from each database. From this information, a hypothesized function was created for 3HDT and potential substrates were selected such as dCMP.
BLASTp
Beginning with BLASTp[1], we queried the FASTA sequence for our protein, PDB ID 3hdt. The alignment of our query sequence with the NK superfamily shows a high degree of overlap indicating this protein is likely a member. The importance of this is that the cytidylate kinase family is a member of the NK superfamily, supporting our claim that 3hdt is a cytidylate kinase. Also, the query hits show a couple cytidylate kinase-like family proteins almost completely allining with our protein 3HDT.

BLASTp Alignment showing a hit with cytidylate kinase-like family, part of the NK superfamily.

Query hits 1-2 show proteins with unknown function while 3-6 match a cytidylate kinase-like family protein.
Pfam
We used the FASTA sequence from 3HDT to do a comparative search in Pfam[2] to find similar protein families our protein may belong. Pfam predicted that our protein is part of the cytidylate kinase which was also shown in the BLASTp results.

Pfam query aligned with a cytidylate kinase-like family.
Top domain results from Pfam.
DALI
Two of the top results from our DALI[3] query (both cytidylate kinases) show a high structural resemblance with 3hdt when overlaid in DALI’s viewer. It is also worth noting that the majority of our results from our DALI query consisted of cytidylate kinases. This information helped our decision to choose a function like cytidylate kinase because of similar alignment between the proteins (structure of the protein = function) indicating similarity in function and our protein relates to them.
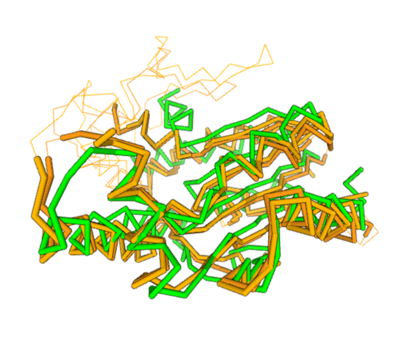
Spatial alignment of putative kinase 3HDT (green) with two cytidylate kinases: 7L4A (dark brown) and 1KDO (light brown).
Docking
We used POCASA to determine potential binding pockets within our protein and PyRx[4] to actually bind dCMP to 3HDT. Then PyMOL was used to visualize the binding pockets and dCMP in the protein. This (in purple) is a potential pocket the substrate dCMP may bind to in the protein, 3HDT. However, this area was where dCMP binded with the highest affinity in PyRx. The (Gly21, Ser22, Gly23, Val27, Thr 142, Gln149, Arg150, Thr197, Leu200, Thr201) interact within that area to help hold the substrate in place in the protein with hydrophobic interactions. Also, there are two amino acids that interact with dCMP with hydrogen bonds. However, these results may not be as accurate because during the docking process on PyRx, we were unsuccessful in getting the program to recognize where the ATP was bonded in the protein when docking our substrate.
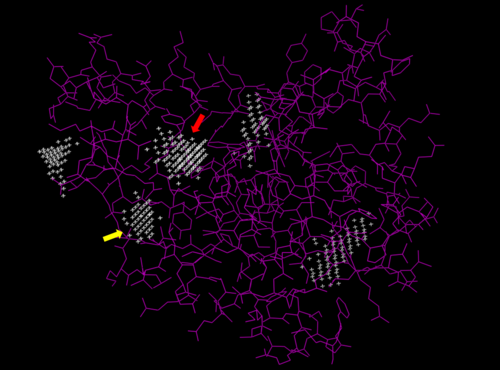
Predicted binding pockets for 3HDT represented by the white stippling.
Some of the predicted bind pockets were areas where ATP (red arrow) and dCMP (yellow arrow) binded to with the highest affinity in PyRx.
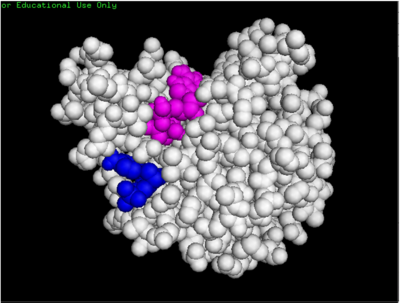
dCMP (purple) docked with 3HDT and cofactor ATP (pink). Visualized in PyMOL
[5].
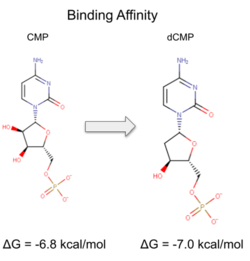
Binding affinity was increased when hydroxyl group removed from ribose ring on CMP to make dCMP
Active site containing dCMP and cofactor ATP can be found with residues important in substrate binding highlighted in purple (Lys146) and yellow (Leu202).
Laboratory Experiments
Coupled kinase assay
Two rounds of coupled kinase assays were run using 3HDT with ATP and dCMP as substrates. The concentration of dCMP was 109mM. The first round of assay (3 total assays) used 5μL of 3HDT and various amounts of 109mM of dCMP. 6.88μL of dCMP resulted in the highest specific activity (0.37669 U/mg) and increasing the substrate amount ~2μL had a similar but slightly less specific activity of 0.3268 U/mg. However, when we repeated the first kinase assays we did (using 5μL of 3HDT), there are discrepancies in the results indicating a potential experimental error such as not pipetting up and down to mix, bubbles, taking too long between mixing the substrate in and reading the plate. In addition, the protein in the second round was older (original protein but about a week old from when it was made) which could have affected the specific activity with the substrate because the protein was starting to expire/decrease function.
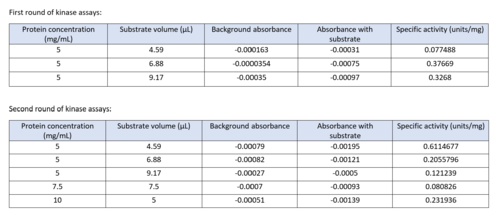
Table of data showing all eight coupled kinase assays with 3HDT
Coupled kinase assay diagram (left) with enzymes shown in color and phosphates in yellow. Phosphorylation of dCMP is measured indirectly through the conversion of NADH to NAD+. Background hydrolysis of NADH is measured and subtracted from the conversion rate in the presence of dCMP to produce specific activities (right).
SDS-PAGE
SDS-PAGE results for the purified protein 3HDT. The total weight of this protein is around 25.79 kD. The first lane (left) contains a size standard. The band in the second lane (right) at ~70kD is not the protein of interest (3HDT) but contains a binding metal protein. There is also a faint band around ~26kD, indicating our protein of interest was present.

Results from running an SDS-PAGE with the purified 3HDT
Conclusion/Future Experiments
Our protein was confirmed to be 3HDT using SDS-PAGE and showed activity during coupled kinase assays. While this confirms that 3HDT is a kinase, the true substrate, however, was likely not dCMP. Further research should be done with molecules such as TMP and GMP in the future to narrow down potential nucleotide substrates or elucidate other types of compounds to be considered as ligands for 3HDT. Also, if we had more time, we would repeat the protein purification process to try to get a higher protein concentration than what we achieved.










