Human Acetylcholinesterase
From Proteopedia
| (10 intermediate revisions not shown.) | |||
| Line 1: | Line 1: | ||
| - | = | + | <StructureSection load='' size='450' side='right' scene='49/497015/Cv/1' caption='Structure of human AChE (PDB code [[4ey4]])'> |
| - | + | The human acetylcholinesterase (AChE) is an enzyme which hydrolyses the neurotransmitter acetylcholine (ACh) in the neuromuscular junctions and in other cholinergic synapses to terminate the neuronal signal. | |
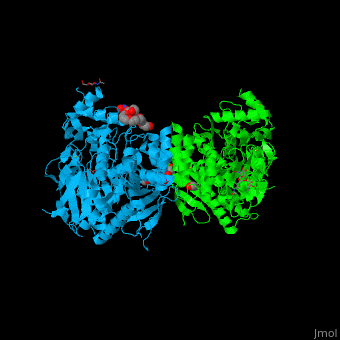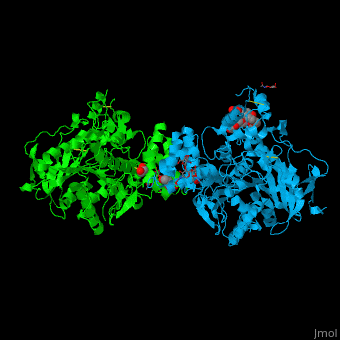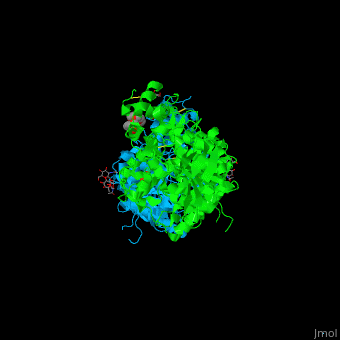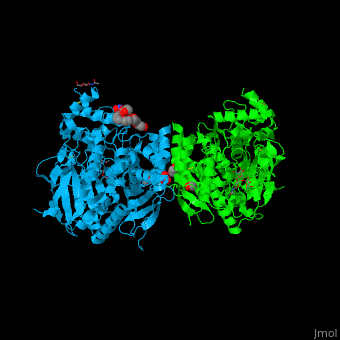
| - | The human acetylcholinesterase (AChE) is an enzyme which hydrolyses the neurotransmitter | + | It has an ellipsoidal shape with dimensions ~ 45Å x 60Å x 65Å. This protein is composed of 531 residues. It consists of 12-stranded, central mixed <scene name='49/497015/Cv/2'>β-sheet</scene> surrounded by 14 <scene name='49/497015/Cv/3'>α helices</scene>. It is a member of the α/β hydrolase fold family<ref>PMID 1409539</ref>. |
| - | It has an ellipsoidal shape with dimensions ~ | + | |
In the physiological conditions, AChE exists as tetramers associated with either collagen-like Q subunit (ColQ) or proline-rich membrane-anchoring protein (PRiMA). The AChE is linked with these anchoring molecules by a "tryptophan amphiphilic tetramerization" domain (WAT). There is also a monomeric form which is soluble in the blood. | In the physiological conditions, AChE exists as tetramers associated with either collagen-like Q subunit (ColQ) or proline-rich membrane-anchoring protein (PRiMA). The AChE is linked with these anchoring molecules by a "tryptophan amphiphilic tetramerization" domain (WAT). There is also a monomeric form which is soluble in the blood. | ||
| Line 15: | Line 14: | ||
The catalytic site of AChE consists of two subsites: the "esteratic" site and "the anionic" site. | The catalytic site of AChE consists of two subsites: the "esteratic" site and "the anionic" site. | ||
| - | In the "esteratic site" a catalytic triad consisting of <scene name=' | + | In the "esteratic site" a catalytic triad consisting of <scene name='49/497015/Cv/7'>E334, H447, S203</scene> forms a planar array that resembles the catalytic triad of serine proteases. |
S203 is activated (it becomes nucleophilic) by E334 and H447. This activation allows the following reaction: the acylation between hydroxyl group of S203 and ACh oxygen (or other agonists). A covalent bond between the enzyme and the substrate creates an oxyanion. This oxyanion then reacts with two glycins setting up a hydrogen bond. | S203 is activated (it becomes nucleophilic) by E334 and H447. This activation allows the following reaction: the acylation between hydroxyl group of S203 and ACh oxygen (or other agonists). A covalent bond between the enzyme and the substrate creates an oxyanion. This oxyanion then reacts with two glycins setting up a hydrogen bond. | ||
| - | In the "anionic" site, the <scene name=' | + | In the "anionic" site, the <scene name='49/497015/Cv/5'>W86</scene> binds trimethylammonium group of ACh. |
Further to these steps the substrate is well positioned to be hydrolysed into acetic acid and cholin. | Further to these steps the substrate is well positioned to be hydrolysed into acetic acid and cholin. | ||
| Line 37: | Line 36: | ||
The molecule which has been the most studied is tacrine. A monomer of tacrine binds strongly to the peripheral site, preventing the subtrate from entry. When tacrine is in the dimer shape, it can bind the catalytic and peripheral sites of AChE. | The molecule which has been the most studied is tacrine. A monomer of tacrine binds strongly to the peripheral site, preventing the subtrate from entry. When tacrine is in the dimer shape, it can bind the catalytic and peripheral sites of AChE. | ||
| + | = Automated computational design of human enzymes for high bacterial expression and stability <ref>PMID 27425410</ref>= | ||
| + | Upon heterologous overexpression, many proteins misfold or aggregate, thus resulting in low functional yields. Human acetylcholinesterase (hAChE), an enzyme mediating synaptic transmission, is a typical case of a human protein that necessitates mammalian systems to obtain functional expression. Using a novel computational strategy, we designed an AChE variant bearing 51 mutations that improved core packing, surface polarity, and backbone rigidity. This variant expressed at ~2,000-fold higher levels in E. coli compared to wild-type hAChE, and exhibited 20°C higher thermostability with no change in enzymatic properties or in the active-site configuration as determined by crystallography. To demonstrate broad utility, we similarly designed four other human and bacterial proteins. Testing at most three designs per protein, we obtained enhanced stability and/or higher yields of soluble protein in E. coli. Our algorithm requires only a 3D structure and several dozen sequences of naturally occurring homologues, and is available at [http://pross.weizmann.ac.il| http://pross.weizmann.ac.il]. | ||
| - | == | + | <scene name='72/728277/Cv/26'>The structural underpinnings of stabilization in the designed variant dAChE4</scene>. <font color='slateblue'><b>Wild type hAChE is shown in blue</b></font> and <font color='darkorange'><b>51 mutated positions, which are distributed throughout dAChE4, are indicated by orange spheres</b></font>. |
| + | The choice of mutations at Gly416 in hAChE illustrates the role of these two filters (alignment scan and computational mutation scanning) in pruning false positives (see static image below). Position 416 is located on a partially exposed helical surface, where the small and flexible amino acid Gly is likely to destabilize hAChE. Indeed, in the alignment of 5 AChE homologues, Gly is infrequent and His is the most prevalent amino acid. Modeling shows, however, that in the specific context of hAChE, His adopts a strained side-chain conformation; in contrast, Gln, the third most prevalent amino acid, is predicted to be most stabilizing owing to its high helical propensity and favorable hydrogen-bonding with Tyr504. The combined filter therefore favors Gln over His for downstream design calculations. | ||
| + | |||
| + | [[Image:MC1.png|left|450px|thumb|Eliminating potentially destabilizing mutations through homologous-sequence analysis and computational mutation scanning. Left: Sequence logo for hAChE position Gly416. The height of letters represents the respective amino acid’s frequency in an alignment of homologous AChE sequences. The evolutionarily ‘allowed’ sequence space (PSSM scores ≥0) at position 416 includes the 9 amino acids shown. Right: Structural models of mutations to the evolutionarily favored amino acid His, and to Gln, which is favored by Rosetta energy calculations. The His side chain is strained due to its proximity to the bulky Tyr504 aromatic ring, whereas the Gln side chain is relaxed and forms a favorable hydrogen bond with Tyr504 (dashed line)]] | ||
| + | {{Clear}} | ||
| + | Scenes highlight stabilizing effects of <scene name='72/728277/Cv/10'>selected mutations</scene> (in <font color='red'><b>red</b></font>), <font color='cyan'><b>wild type hAChE is shown in cyan</b></font> and <font color='lime'><b>designed hAChE is in green</b></font>: | ||
| + | *<scene name='72/728277/Cv/27'>Buried hydrogen bonds</scene>. | ||
| + | *<scene name='72/728277/Cv/28'>Surface polarity</scene>. | ||
| + | *<scene name='72/728277/Cv/29'>Helix capping</scene>. | ||
| + | *<scene name='72/728277/Cv/30'>Loop rigidity</scene>. | ||
| + | *<scene name='72/728277/Cv/31'>Core packing</scene>. | ||
| + | |||
| + | <scene name='72/728277/Cv/32'>Sub-Ångstrom accuracy in alignment of the catalytic triad Ser203, Glu334, and His447</scene> in the crystallographic structure of <font color='gold'><b>dAChE4 (PDB entry: [[5hq3]], yellow)</b></font> compared to <font color='lime'><b>hAChE (PDB entry: [[4ey4]], green)</b></font>. | ||
| + | |||
| + | <scene name='72/728277/Cv/33'>Sub-Ångstrom accuracy in alignment of key residues in the vicinity of the catalytic triad</scene>. | ||
| + | |||
| + | |||
| + | Comparison of the <font color='gold'><b>dAChE4 design model (yellow)</b></font> with the <font color='lime'><b>solved crystal structure (PDB entry: [[5hq3]], green)</b></font> and <font color='violet'><b>(wild-type hAChE (PDB entry: [[4ey4]], violet)</b></font>: | ||
| + | |||
| + | *<scene name='72/728277/Cv/14'>Overall alignment</scene>. | ||
| + | *<scene name='72/728277/Cv/13'>Sub-Ångstrom accuracy in design of 2 small-to-large core mutations</scene>. | ||
| + | *<scene name='72/728277/Cv/19'>The maximal deviation observed between any respective Cα atoms</scene> in the model and structure is 3.1 Å (dashed line). This conformation change likely results from elimination of a side chain-backbone hydrogen bond between Thr112 and Ser110 due to the designed Thr112Ala mutation. | ||
| + | *<scene name='72/728277/Cv/16'>Comparison of designed buried hydrogen bonds</scene>. Val331Asn was predicted to form a hydrogen bond with Glu450 and another with Pro446 in the designed model; in the crystal structure, instead, Asn331 interacts with Glu334 and Glu450. | ||
| + | *<scene name='72/728277/Cv/18'>Leu394Asn forms 2 hydrogen bonds with Pro388 and Asp390, as designed</scene>. | ||
| + | </StructureSection> | ||
| + | |||
| + | == References, for further information on Acetylcholinesterase == | ||
| + | <references/> | ||
'''To the structures used here:''' | '''To the structures used here:''' | ||
Current revision
| |||||||||||



References, for further information on Acetylcholinesterase
- ↑ Ollis DL, Cheah E, Cygler M, Dijkstra B, Frolow F, Franken SM, Harel M, Remington SJ, Silman I, Schrag J, et al.. The alpha/beta hydrolase fold. Protein Eng. 1992 Apr;5(3):197-211. PMID:1409539
- ↑ Goldenzweig A, Goldsmith M, Hill SE, Gertman O, Laurino P, Ashani Y, Dym O, Unger T, Albeck S, Prilusky J, Lieberman RL, Aharoni A, Silman I, Sussman JL, Tawfik DS, Fleishman SJ. Automated Structure- and Sequence-Based Design of Proteins for High Bacterial Expression and Stability. Mol Cell. 2016 Jul 21;63(2):337-346. doi: 10.1016/j.molcel.2016.06.012. Epub 2016, Jul 14. PMID:27425410 doi:http://dx.doi.org/10.1016/j.molcel.2016.06.012
To the structures used here:
- Li W, Mak M, Jiang H, Wang Q, Pang Y, Chen K & Han Y (2009) "Novel anti-Alzheimer's dimer Bis(7)-cognitin: cellular and molecular mechanisms of neuroprotection through multiple targets", Neurotherapeutics, vol.6, p.187-201.
- Zhang D & McCammon JA (2005) "The association of tetrameric acetylcholinesterase with colQ tail: a block normal mode analysis", PLoS Comput Biology, vol.1, p.484-491.
To the active site of acetylcholinesterase
- Rosenberry TL (2009) "Strategies to resolve the catalytic mechanism of acetylcholinesterase", Journal of Molecular Neuroscience.
- Currently (November 05, 2009), part of the content of this page is inspired from a source: http://www.biochimie.univ-montp2.fr/licence/enzymo/ache/ache.htm
Hélène ERASIMUS, Blandine FAUVEL, Tiphaine Jaeg 17:08, 12 November 2009 (IST)
