User:Nitzan Dubovski/Prion
From Proteopedia
< User:Nitzan Dubovski(Difference between revisions)
| (18 intermediate revisions not shown.) | |||
| Line 20: | Line 20: | ||
The normal form of the protein is called PRPc, while the infectious form is called PRPsc. | The normal form of the protein is called PRPc, while the infectious form is called PRPsc. | ||
There are conformational differences between PRPc and PRPsc: | There are conformational differences between PRPc and PRPsc: | ||
| - | The PRPc is soluble in detergents, sensitive to digestion by Protease | + | The PRPc is soluble in detergents, sensitive to digestion by Protease enzymes, and contains predominantly alpha helices. In contrast, the PRPsc is insoluble, has a resistance to proteolysis, and contains predominantly beta pleated. |
==Structure== | ==Structure== | ||
| Line 32: | Line 32: | ||
===Structure of PRPsc=== | ===Structure of PRPsc=== | ||
The damaged form (PRPsc) is built from the same amino acids but takes a different shape, and contains mainly beta sheets. Little is known about the molecular details of this isoform, however it is accepted that during the conversion of PRPc to PRPsc the β-strand content increases substantially, and this altered structure is insoluble, extremely stable, and has a resistance to denaturation by chemical and physical agents. All these features encourage accumulations (known as amyloids) in infected tissues and therefore tissue damage and cell death. | The damaged form (PRPsc) is built from the same amino acids but takes a different shape, and contains mainly beta sheets. Little is known about the molecular details of this isoform, however it is accepted that during the conversion of PRPc to PRPsc the β-strand content increases substantially, and this altered structure is insoluble, extremely stable, and has a resistance to denaturation by chemical and physical agents. All these features encourage accumulations (known as amyloids) in infected tissues and therefore tissue damage and cell death. | ||
| - | During the formation of | + | |
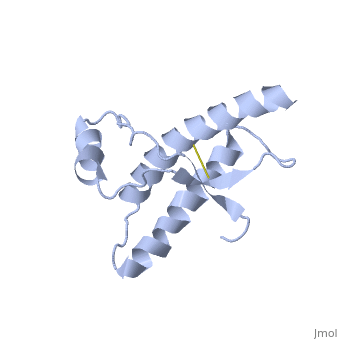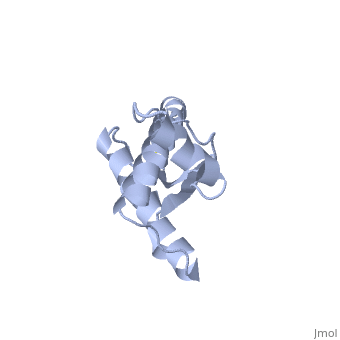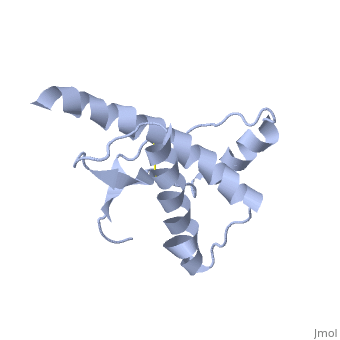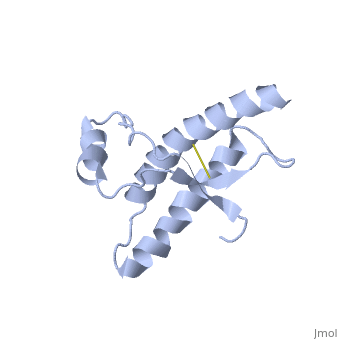
| - | + | During the formation of amyloid and non-amyloid aggregations from recombinant PRPc in vitro, <scene name='68/684796/1qlx/6'>a2 and a3 helices</scene> have been shown to undergo a vest structural rearrangement into a <scene name='68/684796/Prpsc/1'>cross beta-sheet architecture</scene>. Recent evidence suggests a similar conformational change in this region of the protein upon conversion to the PRPsc isoform. The <scene name='68/684796/1qlx/6'>a2 and a3 helices of the PRPc</scene> are being converted into <scene name='68/684796/Prpsc/3'>two discontinuous segments</scene>, that are covalently linked by a disulfide bond between <scene name='68/684796/Prpsc/6'>Cys179 and Cys214</scene>. Then, specific hydrogen bonding interactions between exposed beta strands facilities the formation of a <scene name='68/684796/Prpsc/7'>large molecular weight aggregate</scene>, which is typical for amyloid. The assembly of these fragments into the hexamer is driven by the maximization of hydrogen bonding and burial of hydrophobic side-chains.<ref>Apostol MI et al. Crystal structure of a human prion protein fragment reveals a motif for oligomer formation. 2013</ref> | |
| - | The a2 and a3 helices of the PRPc are | + | |
===Mutations=== | ===Mutations=== | ||
Mutations in the Prion Protein Gene (PRPN) are known to encourage the spontaneous generation of the damaged form- PRPsc. | Mutations in the Prion Protein Gene (PRPN) are known to encourage the spontaneous generation of the damaged form- PRPsc. | ||
| - | The NMR structure of the recombinant human | + | The NMR structure of the recombinant human PRPc from residue 90 to 237 carrying the Q212P, is being presented <scene name='68/684796/Mut/5'>here</scene>. The mutations is caused as a result of glutamine-prolin <scene name='68/684796/Mut/6'>substitution in residue 212</scene>. This amino acid residue is located in the middle of a3 in close proximity to the disulfide bond. This mutation is believed to cause GSS syndrome (a familial prion disease). There are two main differences between <scene name='68/684796/Mut/8'>Q212P structure</scene> and the WT structure. First of all, there are four alpha helices (instead of three), as a result of a small rotation in a3 derive from interruption by Glu221 and Ser222. The break into 2 different helices results in dramatic changes in hydrophobic interactions between a3 and b2-a2 loop region. As <scene name='68/684796/Mut/11'>Tyr225 is unable to contact with Met166</scene>, hydrophobic cluster is opened and accessible to solvent and to hypothetical facilitator of prion conversion. The other remarkable difference is the shape of b2-a2 loop region, as a result of the changes in the hydrophobic interactions. <ref>Ilc G et al. NMR structure of the human prion protein with the pathological Q212P mutation reveals unique structural features. 2010</ref> |
== Function == | == Function == | ||
| - | The cellular normal prion is found in | + | The cellular normal prion is found in various tissues and cell types, but it most highly found in central nervous system <ref>Acevedo-Morantes CY et al. The structure of human prions: from biology to structural models-considerations and pitfalls. 2014</ref>. the PRPc is expressed on the membranes of cells, and it prevents neuronal dysfunction, by using its anti-oxidant activity, and its ability to bind with copper. The PRPc has also been reported to play an important role in cell-cell adhesion, and may therefore be involved in cell-cell communication in the brain. |
The misfolded prion causes the prion diseases- a group of progressive, neurodegenerative disorders <ref>Acevedo-Morantes CY et al. The structure of human prions: from biology to structural models-considerations and pitfalls. 2014</ref>. | The misfolded prion causes the prion diseases- a group of progressive, neurodegenerative disorders <ref>Acevedo-Morantes CY et al. The structure of human prions: from biology to structural models-considerations and pitfalls. 2014</ref>. | ||
| Line 60: | Line 59: | ||
'''Genetic-''' This condition can be inherited. in this case it can also be called familial CJD. | '''Genetic-''' This condition can be inherited. in this case it can also be called familial CJD. | ||
| - | '''Sporadic-''' Sporadic CJD develops suddenly without any known risk factors, because of a spontaneous mutation in the Prion Protein Gene (PRNP). These mutations may lead to changes in the secondary and teritary structure of the PRPc, and could result in the appearance of PRPsc | + | '''Sporadic-''' Sporadic CJD develops suddenly without any known risk factors, because of a spontaneous mutation in the Prion Protein Gene (PRNP). These mutations may lead to changes in the secondary and teritary structure of the PRPc, and could result in the appearance of PRPsc conformers (viruses 2014). Most cases of CJD are sporadic and tend to strike people around age 60. |
'''Acquired-''' The Variant CJD is an infectious type of the disease that is related to “mad cow disease”. Eating infectious meat may cause the disease in humans. The meat may cause normal human prion protein to develop abnormally. The disease is also thought to have been spread to people receiving transplants from infected donors and from contaminated medical equipment. This type of the disease usually affects younger people. | '''Acquired-''' The Variant CJD is an infectious type of the disease that is related to “mad cow disease”. Eating infectious meat may cause the disease in humans. The meat may cause normal human prion protein to develop abnormally. The disease is also thought to have been spread to people receiving transplants from infected donors and from contaminated medical equipment. This type of the disease usually affects younger people. | ||
Current revision
Prion Protein
| |||||||||||