This old version of Proteopedia is provided for student assignments while the new version is undergoing repairs. Content and edits done in this old version of Proteopedia after March 1, 2026 will eventually be lost when it is retired in about June of 2026.
Apply for new accounts at the new Proteopedia. Your logins will work in both the old and new versions.
User:Alisha, Deepa, Pamiz/Sandbox 1
From Proteopedia
(→H+/K+-ATPase) |
(→Conformational Change and Signaling Pathway of H+/K+-ATPase) |
||
| Line 15: | Line 15: | ||
== Conformational Change and Signaling Pathway of H+/K+-ATPase == | == Conformational Change and Signaling Pathway of H+/K+-ATPase == | ||
| - | Inorganic Pi produced from the hydrolysis of ATP drives a conformational change in the enzyme and allows release of H+ into the highly acidic environment [11]. The enzyme catalyzes this reaction by changing conformation states between E1 and E2 [12]. [[Image:e1 to e2.gif|300px|right|thumb| The reaction begins when a hydronium ion binds to the enzyme on the cytoplasmic surface [12]. MgATP will phosphorylate the enzyme at an Asp386 residue in a DKTG amino acid sequence to form the E1~Pi H+ intermediate [12]. E1 undergoes a conformational change to form E2, where the ion site is exposed and H+ is released at a pH ~ 1.0 [7]. Extracellular K+ ions then bind to the same exposed region and the enzyme dephosphorylates [7]. An occluded form of the enzyme (trapped) is formed once K+ ions bind; the enzyme de-occludes, reforms the E1 complex, and K+ is released [7].]] H+/K+-ATPase pumps are normally inactive and inside vesicles [13]. These vesicles are activated through a signaling pathway. | + | Inorganic Pi produced from the hydrolysis of ATP drives a conformational change in the enzyme and allows release of H+ into the highly acidic environment [11]. The enzyme catalyzes this reaction by changing conformation states between E1 and E2 [12]. [[Image:e1 to e2.gif|300px|right|thumb|'''Conformational Change.''' The reaction begins when a hydronium ion binds to the enzyme on the cytoplasmic surface [12]. MgATP will phosphorylate the enzyme at an Asp386 residue in a DKTG amino acid sequence to form the E1~Pi H+ intermediate [12]. E1 undergoes a conformational change to form E2, where the ion site is exposed and H+ is released at a pH ~ 1.0 [7]. Extracellular K+ ions then bind to the same exposed region and the enzyme dephosphorylates [7]. An occluded form of the enzyme (trapped) is formed once K+ ions bind; the enzyme de-occludes, reforms the E1 complex, and K+ is released [7].]] H+/K+-ATPase pumps are normally inactive and inside vesicles [13]. These vesicles are activated through a signaling pathway. |
H+/K+-ATPase signal pathway (acetylcholine, histamine, and gastrin) activates the pump in order to move the vesicles toward the lumen [13]. These signals bind to their corresponding receptors and activate the cAMP and Ca2+ dependent pathways [13]. Increased levels of intracellular Ca2+ and cAMP will promote the translocation of vesicles to the canalicular membrane, activating the H+/K+-ATPase [13]. Histamine binds to Histamine H2, and sends a signal through a G protein which activates adenylate cyclase, and catalyzes the conversion of ATP to cAMP [13].Gastrin will stimulate the release of histamine by binding to CCK2 [13]. Acetylcholine binds to Muscarinic M3 and releases Ca2+ from the endoplasmic reticulum [13]. | H+/K+-ATPase signal pathway (acetylcholine, histamine, and gastrin) activates the pump in order to move the vesicles toward the lumen [13]. These signals bind to their corresponding receptors and activate the cAMP and Ca2+ dependent pathways [13]. Increased levels of intracellular Ca2+ and cAMP will promote the translocation of vesicles to the canalicular membrane, activating the H+/K+-ATPase [13]. Histamine binds to Histamine H2, and sends a signal through a G protein which activates adenylate cyclase, and catalyzes the conversion of ATP to cAMP [13].Gastrin will stimulate the release of histamine by binding to CCK2 [13]. Acetylcholine binds to Muscarinic M3 and releases Ca2+ from the endoplasmic reticulum [13]. | ||
| - | + | [[Image:signaling pathway 2.gif]] '''Acetylcholine, Histamine, Gastrin Signals''' [14] | |
== Esomeprazole Mechanism == | == Esomeprazole Mechanism == | ||
Revision as of 20:44, 6 December 2013
Contents |
Introduction
![2D Structure & Biochemical Parameters of Esomeprazole Esomeprazole has two important pyridine and benzimidazole moieties linked through a methylenesulfinyl group [3]. pKa, IC50, AUC, Cmax, and half life values of Esomeprazole [4,5].](/wiki/index.php/Image:Eso.jpg)
Ulcers caused by the bacterium Helicobacter pylori can be treated using Esomeprazole in conjunction with proper antibiotics [1]. Gastric acid is released through the H+/K+-ATPase pump, which is the final step in acid release [2]. Esomeprazole is a specific, irreversible inhibitor of the pump [2].
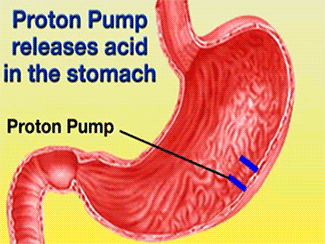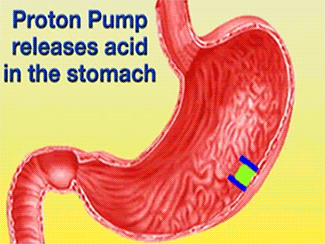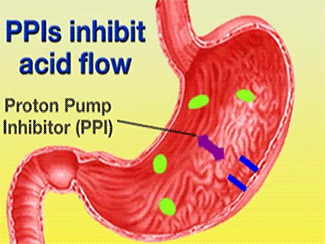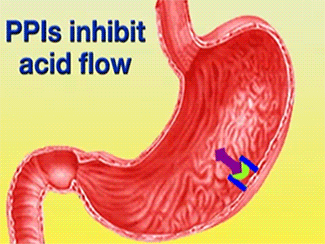 Function of PPIs[6]
Function of PPIs[6]

H+/K+-ATPase
The H+/K+-ATPase pump is part of the P-type ATPase family located within the cytoplasmic membrane of resting parietal cells; powered through ATP hydrolysis, the ATPase is translocated to the canalicular membrane and begins to pump cytoplasmic H+ into the canalicular space, in exchange for extracellular K+ ions. H+/K+-ATPase is an α,β-heterodimeric enzyme, where the catalytic site is present in the α subunit [7]. Transmembrane segments TM4, TM5, TM6, and TM8, are located in the α subunit and contain the ion binding region of the enzyme [7]. Binding of ions and ATP to these domains will induce movements in the membrane domain that catalyze ion displacement [7]. Targeting this enzyme using PPIs is the most effective therapeutic control agent of gastric acid secretion [8].
Parietal Cell Acid Secretion [9]
Inhibition of H+/K+ ATPase [10]


Conformational Change and Signaling Pathway of H+/K+-ATPase
Inorganic Pi produced from the hydrolysis of ATP drives a conformational change in the enzyme and allows release of H+ into the highly acidic environment [11]. The enzyme catalyzes this reaction by changing conformation states between E1 and E2 [12].![Conformational Change. The reaction begins when a hydronium ion binds to the enzyme on the cytoplasmic surface [12]. MgATP will phosphorylate the enzyme at an Asp386 residue in a DKTG amino acid sequence to form the E1~Pi H+ intermediate [12]. E1 undergoes a conformational change to form E2, where the ion site is exposed and H+ is released at a pH ~ 1.0 [7]. Extracellular K+ ions then bind to the same exposed region and the enzyme dephosphorylates [7]. An occluded form of the enzyme (trapped) is formed once K+ ions bind; the enzyme de-occludes, reforms the E1 complex, and K+ is released [7].](/wiki/index.php/Image:E1_to_e2.gif)
H+/K+-ATPase signal pathway (acetylcholine, histamine, and gastrin) activates the pump in order to move the vesicles toward the lumen [13]. These signals bind to their corresponding receptors and activate the cAMP and Ca2+ dependent pathways [13]. Increased levels of intracellular Ca2+ and cAMP will promote the translocation of vesicles to the canalicular membrane, activating the H+/K+-ATPase [13]. Histamine binds to Histamine H2, and sends a signal through a G protein which activates adenylate cyclase, and catalyzes the conversion of ATP to cAMP [13].Gastrin will stimulate the release of histamine by binding to CCK2 [13]. Acetylcholine binds to Muscarinic M3 and releases Ca2+ from the endoplasmic reticulum [13].
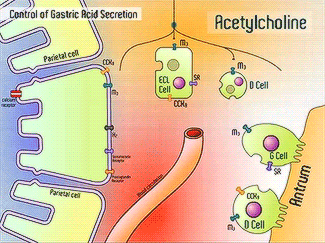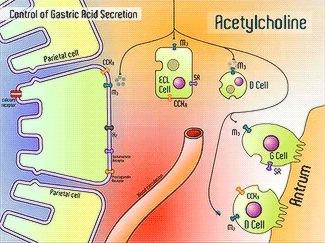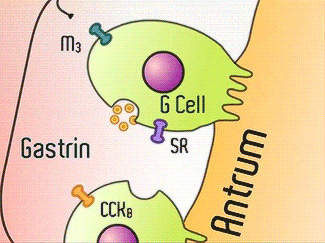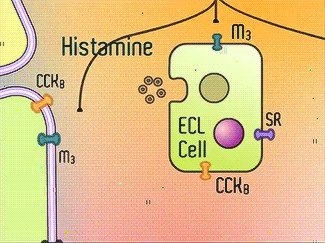 Acetylcholine, Histamine, Gastrin Signals [14]
Acetylcholine, Histamine, Gastrin Signals [14]

Esomeprazole Mechanism
Esomeprazole is protonated twice within the acidic environment, and forms the active inhibitor—achiral sulfenamide at pKa of 1.0 [15].![Activation of Esomeprazole to sulfonamide [7]. R1=OCH3, R2=CH3, R3=CH3, R4=CH3, X=CH, Bz=benzimidazole, Py=pyridine[7]. Mechanism: (1) protonation of Py, (2) protonation of Bz, (3) intramolecular rearrangement of BzH+-Py, forms sulfenic acid (4) dehydration to form sulfenamide (5) disulfide bond formation between enzyme Cys residues and sulfonamide [7].](/wiki/index.php/Image:Mechanism_of_inhibition.jpg)
Drug-Molecule Interaction
|
Esomeprazole Resistance
Resistance to treatment with PPIs, including Esomeprazole, has been speculated among Barrett’s Esophagus (BE) patients who did not indicate any symptomatic improvement after being placed on a standard PPI drug dose [20]. No contributory mutations causing PPI resistance have been found [20]. It is speculated that the high acid exposure in BE patients may be due to “reflux diathesis” rather than resistance to gastric acid secretion [21]. Other possible reasons for PPI failure include Helicobacter pylori infection, rapid metabolism, and bioavailability; reasons of clinical significance include delayed gastric emptying and visceral hypersensitivity [20]. More studies need to be conducted to understand the mechanisms underlying the development of resistance to PPIs [20].
