This old version of Proteopedia is provided for student assignments while the new version is undergoing repairs. Content and edits done in this old version of Proteopedia after March 1, 2026 will eventually be lost when it is retired in about June of 2026.
Apply for new accounts at the new Proteopedia. Your logins will work in both the old and new versions.
1ktz
From Proteopedia
(Difference between revisions)
| Line 3: | Line 3: | ||
== Structural highlights == | == Structural highlights == | ||
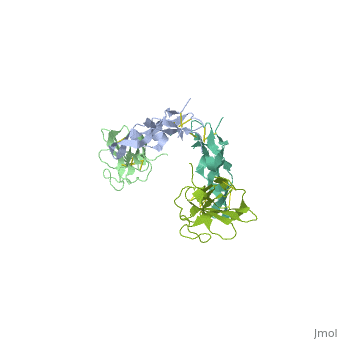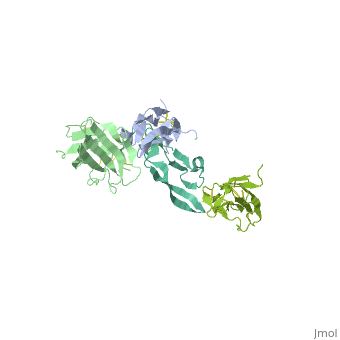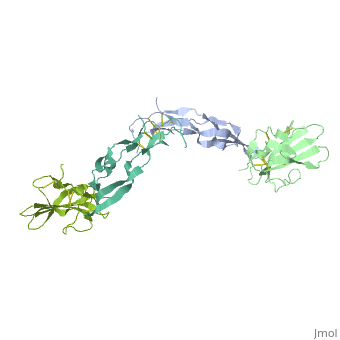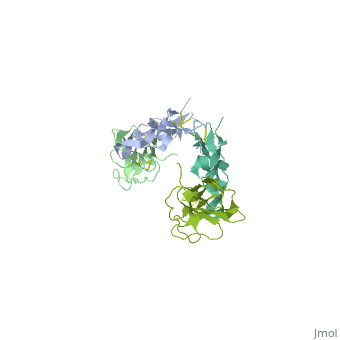
<table><tr><td colspan='2'>[[1ktz]] is a 2 chain structure with sequence from [http://en.wikipedia.org/wiki/Homo_sapiens Homo sapiens]. Full crystallographic information is available from [http://oca.weizmann.ac.il/oca-bin/ocashort?id=1KTZ OCA]. For a <b>guided tour on the structure components</b> use [http://oca.weizmann.ac.il/oca-docs/fgij/fg.htm?mol=1KTZ FirstGlance]. <br> | <table><tr><td colspan='2'>[[1ktz]] is a 2 chain structure with sequence from [http://en.wikipedia.org/wiki/Homo_sapiens Homo sapiens]. Full crystallographic information is available from [http://oca.weizmann.ac.il/oca-bin/ocashort?id=1KTZ OCA]. For a <b>guided tour on the structure components</b> use [http://oca.weizmann.ac.il/oca-docs/fgij/fg.htm?mol=1KTZ FirstGlance]. <br> | ||
| - | </td></tr><tr><td class="sblockLbl"><b>Activity:</b></td><td class="sblockDat"><span class='plainlinks'>[http://en.wikipedia.org/wiki/Non-specific_serine/threonine_protein_kinase Non-specific serine/threonine protein kinase], with EC number [http://www.brenda-enzymes.info/php/result_flat.php4?ecno=2.7.11.1 2.7.11.1] </span></td></tr> | + | </td></tr><tr id='activity'><td class="sblockLbl"><b>Activity:</b></td><td class="sblockDat"><span class='plainlinks'>[http://en.wikipedia.org/wiki/Non-specific_serine/threonine_protein_kinase Non-specific serine/threonine protein kinase], with EC number [http://www.brenda-enzymes.info/php/result_flat.php4?ecno=2.7.11.1 2.7.11.1] </span></td></tr> |
| - | <tr><td class="sblockLbl"><b>Resources:</b></td><td class="sblockDat"><span class='plainlinks'>[http://oca.weizmann.ac.il/oca-docs/fgij/fg.htm?mol=1ktz FirstGlance], [http://oca.weizmann.ac.il/oca-bin/ocaids?id=1ktz OCA], [http://www.rcsb.org/pdb/explore.do?structureId=1ktz RCSB], [http://www.ebi.ac.uk/pdbsum/1ktz PDBsum]</span></td></tr> | + | <tr id='resources'><td class="sblockLbl"><b>Resources:</b></td><td class="sblockDat"><span class='plainlinks'>[http://oca.weizmann.ac.il/oca-docs/fgij/fg.htm?mol=1ktz FirstGlance], [http://oca.weizmann.ac.il/oca-bin/ocaids?id=1ktz OCA], [http://www.rcsb.org/pdb/explore.do?structureId=1ktz RCSB], [http://www.ebi.ac.uk/pdbsum/1ktz PDBsum]</span></td></tr> |
| - | <table> | + | </table> |
== Disease == | == Disease == | ||
[[http://www.uniprot.org/uniprot/TGFB3_HUMAN TGFB3_HUMAN]] Defects in TGFB3 are a cause of familial arrhythmogenic right ventricular dysplasia type 1 (ARVD1) [MIM:[http://omim.org/entry/107970 107970]]; also known as arrhythmogenic right ventricular cardiomyopathy 1 (ARVC1). ARVD is an autosomal dominant disease characterized by partial degeneration of the myocardium of the right ventricle, electrical instability, and sudden death. It is clinically defined by electrocardiographic and angiographic criteria; pathologic findings, replacement of ventricular myocardium with fatty and fibrous elements, preferentially involve the right ventricular free wall.<ref>PMID:15639475</ref> [[http://www.uniprot.org/uniprot/TGFR2_HUMAN TGFR2_HUMAN]] Defects in TGFBR2 are the cause of hereditary non-polyposis colorectal cancer type 6 (HNPCC6) [MIM:[http://omim.org/entry/614331 614331]]. Mutations in more than one gene locus can be involved alone or in combination in the production of the HNPCC phenotype (also called Lynch syndrome). Most families with clinically recognized HNPCC have mutations in either MLH1 or MSH2 genes. HNPCC is an autosomal, dominantly inherited disease associated with marked increase in cancer susceptibility. It is characterized by a familial predisposition to early onset colorectal carcinoma (CRC) and extra-colonic cancers of the gastrointestinal, urological and female reproductive tracts. HNPCC is reported to be the most common form of inherited colorectal cancer in the Western world, and accounts for 15% of all colon cancers. Cancers in HNPCC originate within benign neoplastic polyps termed adenomas. Clinically, HNPCC is often divided into two subgroups. Type I: hereditary predisposition to colorectal cancer, a young age of onset, and carcinoma observed in the proximal colon. Type II: patients have an increased risk for cancers in certain tissues such as the uterus, ovary, breast, stomach, small intestine, skin, and larynx in addition to the colon. Diagnosis of classical HNPCC is based on the Amsterdam criteria: 3 or more relatives affected by colorectal cancer, one a first degree relative of the other two; 2 or more generation affected; 1 or more colorectal cancers presenting before 50 years of age; exclusion of hereditary polyposis syndromes. The term "suspected HNPCC" or "incomplete HNPCC" can be used to describe families who do not or only partially fulfill the Amsterdam criteria, but in whom a genetic basis for colon cancer is strongly suspected. HNPCC6 is a type of colorectal cancer complying with the clinical criteria of HNPCC, except that the onset of cancer was beyond 50 years of age in all cases.<ref>PMID:9590282</ref> Defects in TGFBR2 are a cause of esophageal cancer (ESCR) [MIM:[http://omim.org/entry/133239 133239]]. Defects in TGFBR2 are the cause of Loeys-Dietz syndrome type 1B (LDS1B) [MIM:[http://omim.org/entry/610168 610168]]. LDS1 is an aortic aneurysm syndrome with widespread systemic involvement. The disorder is characterized by arterial tortuosity and aneurysms, craniosynostosis, hypertelorism, and bifid uvula or cleft palate. Other findings include exotropy, micrognathia and retrognathia, structural brain abnormalities, intellectual deficit, congenital heart disease, translucent skin, joint hyperlaxity and aneurysm with dissection throughout the arterial tree.<ref>PMID:15731757</ref> <ref>PMID:16251899</ref> <ref>PMID:20101701</ref> <ref>PMID:20358619</ref> <ref>PMID:22113417</ref> <ref>PMID:21949523</ref> Defects in TGFBR2 are the cause of Loeys-Dietz syndrome type 2B (LDS2B) [MIM:[http://omim.org/entry/610380 610380]]. An aortic aneurysm syndrome with widespread systemic involvement. Physical findings include prominent joint laxity, easy bruising, wide and atrophic scars, velvety and translucent skin with easily visible veins, spontaneous rupture of the spleen or bowel, diffuse arterial aneurysms and dissections, and catastrophic complications of pregnancy, including rupture of the gravid uterus and the arteries, either during pregnancy or in the immediate postpartum period. LDS2 is characterized by the absence of craniofacial abnormalities with the exception of bifid uvula that can be present in some patients. Note=TGFBR2 mutations Cys-460 and His-460 have been reported to be associated with thoracic aortic aneurysms and dissection (TAAD). This phenotype, also known as thoracic aortic aneurysms type 3 (AAT3), is distinguised from LDS2B by having aneurysms restricted to thoracic aorta. As individuals carrying these mutations also exhibit descending aortic disease and aneurysms of other arteries (PubMed:16027248), they have been considered as LDS2B by the OMIM resource. | [[http://www.uniprot.org/uniprot/TGFB3_HUMAN TGFB3_HUMAN]] Defects in TGFB3 are a cause of familial arrhythmogenic right ventricular dysplasia type 1 (ARVD1) [MIM:[http://omim.org/entry/107970 107970]]; also known as arrhythmogenic right ventricular cardiomyopathy 1 (ARVC1). ARVD is an autosomal dominant disease characterized by partial degeneration of the myocardium of the right ventricle, electrical instability, and sudden death. It is clinically defined by electrocardiographic and angiographic criteria; pathologic findings, replacement of ventricular myocardium with fatty and fibrous elements, preferentially involve the right ventricular free wall.<ref>PMID:15639475</ref> [[http://www.uniprot.org/uniprot/TGFR2_HUMAN TGFR2_HUMAN]] Defects in TGFBR2 are the cause of hereditary non-polyposis colorectal cancer type 6 (HNPCC6) [MIM:[http://omim.org/entry/614331 614331]]. Mutations in more than one gene locus can be involved alone or in combination in the production of the HNPCC phenotype (also called Lynch syndrome). Most families with clinically recognized HNPCC have mutations in either MLH1 or MSH2 genes. HNPCC is an autosomal, dominantly inherited disease associated with marked increase in cancer susceptibility. It is characterized by a familial predisposition to early onset colorectal carcinoma (CRC) and extra-colonic cancers of the gastrointestinal, urological and female reproductive tracts. HNPCC is reported to be the most common form of inherited colorectal cancer in the Western world, and accounts for 15% of all colon cancers. Cancers in HNPCC originate within benign neoplastic polyps termed adenomas. Clinically, HNPCC is often divided into two subgroups. Type I: hereditary predisposition to colorectal cancer, a young age of onset, and carcinoma observed in the proximal colon. Type II: patients have an increased risk for cancers in certain tissues such as the uterus, ovary, breast, stomach, small intestine, skin, and larynx in addition to the colon. Diagnosis of classical HNPCC is based on the Amsterdam criteria: 3 or more relatives affected by colorectal cancer, one a first degree relative of the other two; 2 or more generation affected; 1 or more colorectal cancers presenting before 50 years of age; exclusion of hereditary polyposis syndromes. The term "suspected HNPCC" or "incomplete HNPCC" can be used to describe families who do not or only partially fulfill the Amsterdam criteria, but in whom a genetic basis for colon cancer is strongly suspected. HNPCC6 is a type of colorectal cancer complying with the clinical criteria of HNPCC, except that the onset of cancer was beyond 50 years of age in all cases.<ref>PMID:9590282</ref> Defects in TGFBR2 are a cause of esophageal cancer (ESCR) [MIM:[http://omim.org/entry/133239 133239]]. Defects in TGFBR2 are the cause of Loeys-Dietz syndrome type 1B (LDS1B) [MIM:[http://omim.org/entry/610168 610168]]. LDS1 is an aortic aneurysm syndrome with widespread systemic involvement. The disorder is characterized by arterial tortuosity and aneurysms, craniosynostosis, hypertelorism, and bifid uvula or cleft palate. Other findings include exotropy, micrognathia and retrognathia, structural brain abnormalities, intellectual deficit, congenital heart disease, translucent skin, joint hyperlaxity and aneurysm with dissection throughout the arterial tree.<ref>PMID:15731757</ref> <ref>PMID:16251899</ref> <ref>PMID:20101701</ref> <ref>PMID:20358619</ref> <ref>PMID:22113417</ref> <ref>PMID:21949523</ref> Defects in TGFBR2 are the cause of Loeys-Dietz syndrome type 2B (LDS2B) [MIM:[http://omim.org/entry/610380 610380]]. An aortic aneurysm syndrome with widespread systemic involvement. Physical findings include prominent joint laxity, easy bruising, wide and atrophic scars, velvety and translucent skin with easily visible veins, spontaneous rupture of the spleen or bowel, diffuse arterial aneurysms and dissections, and catastrophic complications of pregnancy, including rupture of the gravid uterus and the arteries, either during pregnancy or in the immediate postpartum period. LDS2 is characterized by the absence of craniofacial abnormalities with the exception of bifid uvula that can be present in some patients. Note=TGFBR2 mutations Cys-460 and His-460 have been reported to be associated with thoracic aortic aneurysms and dissection (TAAD). This phenotype, also known as thoracic aortic aneurysms type 3 (AAT3), is distinguised from LDS2B by having aneurysms restricted to thoracic aorta. As individuals carrying these mutations also exhibit descending aortic disease and aneurysms of other arteries (PubMed:16027248), they have been considered as LDS2B by the OMIM resource. | ||
| Line 34: | Line 34: | ||
[[Category: Homo sapiens]] | [[Category: Homo sapiens]] | ||
[[Category: Non-specific serine/threonine protein kinase]] | [[Category: Non-specific serine/threonine protein kinase]] | ||
| - | [[Category: Deep, S | + | [[Category: Deep, S]] |
| - | [[Category: Hart, P J | + | [[Category: Hart, P J]] |
| - | [[Category: Hinck, A P | + | [[Category: Hinck, A P]] |
| - | [[Category: Hinck, C S | + | [[Category: Hinck, C S]] |
| - | [[Category: Shu, Z | + | [[Category: Shu, Z]] |
| - | [[Category: Taylor, A B | + | [[Category: Taylor, A B]] |
[[Category: Cytokine-cytokine receptor complex]] | [[Category: Cytokine-cytokine receptor complex]] | ||
[[Category: Cytokine-receptor complex]] | [[Category: Cytokine-receptor complex]] | ||
Revision as of 16:02, 5 January 2015
Crystal Structure of the Human TGF-beta Type II Receptor Extracellular Domain in Complex with TGF-beta3
| |||||||||||