We apologize for Proteopedia being slow to respond. For the past two years, a new implementation of Proteopedia has been being built. Soon, it will replace this 18-year old system. All existing content will be moved to the new system at a date that will be announced here.
1ya5
From Proteopedia
(Difference between revisions)
| Line 3: | Line 3: | ||
== Structural highlights == | == Structural highlights == | ||
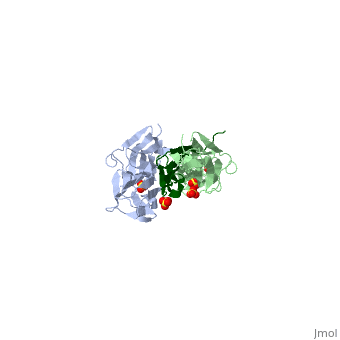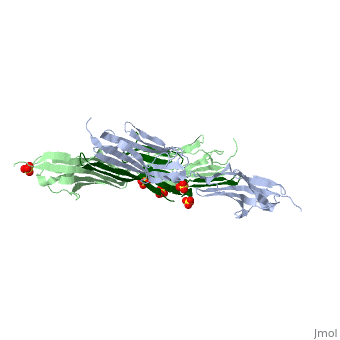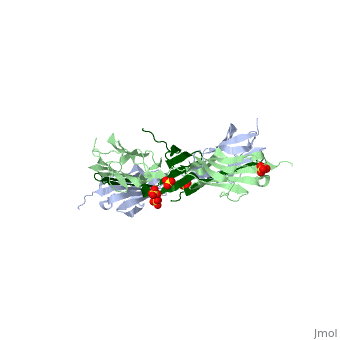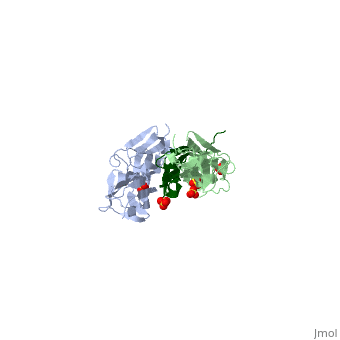
<table><tr><td colspan='2'>[[1ya5]] is a 3 chain structure with sequence from [http://en.wikipedia.org/wiki/Homo_sapiens Homo sapiens]. Full crystallographic information is available from [http://oca.weizmann.ac.il/oca-bin/ocashort?id=1YA5 OCA]. For a <b>guided tour on the structure components</b> use [http://oca.weizmann.ac.il/oca-docs/fgij/fg.htm?mol=1YA5 FirstGlance]. <br> | <table><tr><td colspan='2'>[[1ya5]] is a 3 chain structure with sequence from [http://en.wikipedia.org/wiki/Homo_sapiens Homo sapiens]. Full crystallographic information is available from [http://oca.weizmann.ac.il/oca-bin/ocashort?id=1YA5 OCA]. For a <b>guided tour on the structure components</b> use [http://oca.weizmann.ac.il/oca-docs/fgij/fg.htm?mol=1YA5 FirstGlance]. <br> | ||
| - | </td></tr><tr><td class="sblockLbl"><b>[[Ligand|Ligands:]]</b></td><td class="sblockDat"><scene name='pdbligand=SO4:SULFATE+ION'>SO4</scene>< | + | </td></tr><tr id='ligand'><td class="sblockLbl"><b>[[Ligand|Ligands:]]</b></td><td class="sblockDat"><scene name='pdbligand=SO4:SULFATE+ION'>SO4</scene></td></tr> |
| - | <tr><td class="sblockLbl"><b>Resources:</b></td><td class="sblockDat"><span class='plainlinks'>[http://oca.weizmann.ac.il/oca-docs/fgij/fg.htm?mol=1ya5 FirstGlance], [http://oca.weizmann.ac.il/oca-bin/ocaids?id=1ya5 OCA], [http://www.rcsb.org/pdb/explore.do?structureId=1ya5 RCSB], [http://www.ebi.ac.uk/pdbsum/1ya5 PDBsum]</span></td></tr> | + | <tr id='resources'><td class="sblockLbl"><b>Resources:</b></td><td class="sblockDat"><span class='plainlinks'>[http://oca.weizmann.ac.il/oca-docs/fgij/fg.htm?mol=1ya5 FirstGlance], [http://oca.weizmann.ac.il/oca-bin/ocaids?id=1ya5 OCA], [http://www.rcsb.org/pdb/explore.do?structureId=1ya5 RCSB], [http://www.ebi.ac.uk/pdbsum/1ya5 PDBsum]</span></td></tr> |
| - | <table> | + | </table> |
== Disease == | == Disease == | ||
[[http://www.uniprot.org/uniprot/TITIN_HUMAN TITIN_HUMAN]] Defects in TTN are the cause of hereditary myopathy with early respiratory failure (HMERF) [MIM:[http://omim.org/entry/603689 603689]]; also known as Edstrom myopathy. HMERF is an autosomal dominant, adult-onset myopathy with early respiratory muscle involvement.<ref>PMID:15802564</ref> Defects in TTN are the cause of familial hypertrophic cardiomyopathy type 9 (CMH9) [MIM:[http://omim.org/entry/613765 613765]]. Familial hypertrophic cardiomyopathy is a hereditary heart disorder characterized by ventricular hypertrophy, which is usually asymmetric and often involves the interventricular septum. The symptoms include dyspnea, syncope, collapse, palpitations, and chest pain. They can be readily provoked by exercise. The disorder has inter- and intrafamilial variability ranging from benign to malignant forms with high risk of cardiac failure and sudden cardiac death.<ref>PMID:10462489</ref> Defects in TTN are the cause of cardiomyopathy dilated type 1G (CMD1G) [MIM:[http://omim.org/entry/604145 604145]]. Dilated cardiomyopathy is a disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death.<ref>PMID:11846417</ref> <ref>PMID:11788824</ref> <ref>PMID:16465475</ref> Defects in TTN are the cause of tardive tibial muscular dystrophy (TMD) [MIM:[http://omim.org/entry/600334 600334]]; also known as Udd myopathy. TMD is an autosomal dominant, late-onset distal myopathy. Muscle weakness and atrophy are usually confined to the anterior compartment of the lower leg, in particular the tibialis anterior muscle. Clinical symptoms usually occur at age 35-45 years or much later.<ref>PMID:12145747</ref> <ref>PMID:12891679</ref> Defects in TTN are the cause of limb-girdle muscular dystrophy type 2J (LGMD2J) [MIM:[http://omim.org/entry/608807 608807]]. LGMD2J is an autosomal recessive degenerative myopathy characterized by progressive weakness of the pelvic and shoulder girdle muscles. Severe disability is observed within 20 years of onset. Defects in TTN are the cause of early-onset myopathy with fatal cardiomyopathy (EOMFC) [MIM:[http://omim.org/entry/611705 611705]]. Early-onset myopathies are inherited muscle disorders that manifest typically from birth or infancy with hypotonia, muscle weakness, and delayed motor development. EOMFC is a titinopathy that, in contrast with the previously described examples, involves both heart and skeletal muscle, has a congenital onset, and is purely recessive. This phenotype is due to homozygous out-of-frame TTN deletions, which lead to a total absence of titin's C-terminal end from striated muscles and to secondary CAPN3 depletion.<ref>PMID:17444505</ref> [[http://www.uniprot.org/uniprot/TELT_HUMAN TELT_HUMAN]] Defects in TCAP are a cause of familial hypertrophic cardiomyopathy (CMH) [MIM:[http://omim.org/entry/192600 192600]]; also designated FHC or HCM. Familial hypertrophic cardiomyopathy is a hereditary heart disorder characterized by ventricular hypertrophy, which is usually asymmetric and often involves the interventricular septum. The symptoms include dyspnea, syncope, collapse, palpitations, and chest pain. They can be readily provoked by exercise. The disorder has inter- and intrafamilial variability ranging from benign to malignant forms with high risk of cardiac failure and sudden cardiac death.<ref>PMID:15582318</ref> Defects in TCAP are a cause of limb-girdle muscular dystrophy type 2G (LGMD2G) [MIM:[http://omim.org/entry/601954 601954]]. LGMD2G is an autosomal recessive degenerative myopathy characterized by proximal and distal muscle weakness and atrophy in the limbs, dystrophic changes on muscle biopsy, and absence of telethonin. Cardiac muscle is involved in a subset of patients.<ref>PMID:10655062</ref> Defects in TCAP are the cause of cardiomyopathy dilated type 1N (CMD1N) [MIM:[http://omim.org/entry/607487 607487]]. Dilated cardiomyopathy is a disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death.<ref>PMID:15582318</ref> <ref>PMID:12507422</ref> <ref>PMID:16352453</ref> | [[http://www.uniprot.org/uniprot/TITIN_HUMAN TITIN_HUMAN]] Defects in TTN are the cause of hereditary myopathy with early respiratory failure (HMERF) [MIM:[http://omim.org/entry/603689 603689]]; also known as Edstrom myopathy. HMERF is an autosomal dominant, adult-onset myopathy with early respiratory muscle involvement.<ref>PMID:15802564</ref> Defects in TTN are the cause of familial hypertrophic cardiomyopathy type 9 (CMH9) [MIM:[http://omim.org/entry/613765 613765]]. Familial hypertrophic cardiomyopathy is a hereditary heart disorder characterized by ventricular hypertrophy, which is usually asymmetric and often involves the interventricular septum. The symptoms include dyspnea, syncope, collapse, palpitations, and chest pain. They can be readily provoked by exercise. The disorder has inter- and intrafamilial variability ranging from benign to malignant forms with high risk of cardiac failure and sudden cardiac death.<ref>PMID:10462489</ref> Defects in TTN are the cause of cardiomyopathy dilated type 1G (CMD1G) [MIM:[http://omim.org/entry/604145 604145]]. Dilated cardiomyopathy is a disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death.<ref>PMID:11846417</ref> <ref>PMID:11788824</ref> <ref>PMID:16465475</ref> Defects in TTN are the cause of tardive tibial muscular dystrophy (TMD) [MIM:[http://omim.org/entry/600334 600334]]; also known as Udd myopathy. TMD is an autosomal dominant, late-onset distal myopathy. Muscle weakness and atrophy are usually confined to the anterior compartment of the lower leg, in particular the tibialis anterior muscle. Clinical symptoms usually occur at age 35-45 years or much later.<ref>PMID:12145747</ref> <ref>PMID:12891679</ref> Defects in TTN are the cause of limb-girdle muscular dystrophy type 2J (LGMD2J) [MIM:[http://omim.org/entry/608807 608807]]. LGMD2J is an autosomal recessive degenerative myopathy characterized by progressive weakness of the pelvic and shoulder girdle muscles. Severe disability is observed within 20 years of onset. Defects in TTN are the cause of early-onset myopathy with fatal cardiomyopathy (EOMFC) [MIM:[http://omim.org/entry/611705 611705]]. Early-onset myopathies are inherited muscle disorders that manifest typically from birth or infancy with hypotonia, muscle weakness, and delayed motor development. EOMFC is a titinopathy that, in contrast with the previously described examples, involves both heart and skeletal muscle, has a congenital onset, and is purely recessive. This phenotype is due to homozygous out-of-frame TTN deletions, which lead to a total absence of titin's C-terminal end from striated muscles and to secondary CAPN3 depletion.<ref>PMID:17444505</ref> [[http://www.uniprot.org/uniprot/TELT_HUMAN TELT_HUMAN]] Defects in TCAP are a cause of familial hypertrophic cardiomyopathy (CMH) [MIM:[http://omim.org/entry/192600 192600]]; also designated FHC or HCM. Familial hypertrophic cardiomyopathy is a hereditary heart disorder characterized by ventricular hypertrophy, which is usually asymmetric and often involves the interventricular septum. The symptoms include dyspnea, syncope, collapse, palpitations, and chest pain. They can be readily provoked by exercise. The disorder has inter- and intrafamilial variability ranging from benign to malignant forms with high risk of cardiac failure and sudden cardiac death.<ref>PMID:15582318</ref> Defects in TCAP are a cause of limb-girdle muscular dystrophy type 2G (LGMD2G) [MIM:[http://omim.org/entry/601954 601954]]. LGMD2G is an autosomal recessive degenerative myopathy characterized by proximal and distal muscle weakness and atrophy in the limbs, dystrophic changes on muscle biopsy, and absence of telethonin. Cardiac muscle is involved in a subset of patients.<ref>PMID:10655062</ref> Defects in TCAP are the cause of cardiomyopathy dilated type 1N (CMD1N) [MIM:[http://omim.org/entry/607487 607487]]. Dilated cardiomyopathy is a disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death.<ref>PMID:15582318</ref> <ref>PMID:12507422</ref> <ref>PMID:16352453</ref> | ||
| Line 37: | Line 37: | ||
</StructureSection> | </StructureSection> | ||
[[Category: Homo sapiens]] | [[Category: Homo sapiens]] | ||
| - | [[Category: Pinotsis, N | + | [[Category: Pinotsis, N]] |
| - | [[Category: Popov, A | + | [[Category: Popov, A]] |
| - | [[Category: Wilmanns, M | + | [[Category: Wilmanns, M]] |
| - | [[Category: Zou, P | + | [[Category: Zou, P]] |
[[Category: Ig-like domain]] | [[Category: Ig-like domain]] | ||
[[Category: Structural protein]] | [[Category: Structural protein]] | ||
Revision as of 10:22, 8 January 2015
Crystal structure of the titin domains z1z2 in complex with telethonin
| |||||||||||

Categories: Homo sapiens | Pinotsis, N | Popov, A | Wilmanns, M | Zou, P | Ig-like domain | Structural protein | T-cap | Telethonin | Titin | Z1 | Z2
