|
|
| Line 20: |
Line 20: |
| | Residues <scene name='61/612805/N-c_rainbow/1'>73-326</scene> form a mixed alpha/beta-globular structure, encompassing two independently folded modules corresponding to the B and C domains connected by a flexible linker. | | Residues <scene name='61/612805/N-c_rainbow/1'>73-326</scene> form a mixed alpha/beta-globular structure, encompassing two independently folded modules corresponding to the B and C domains connected by a flexible linker. |
| | | | |
| - | The central B domain <scene name='61/612805/Secondary_structure_b/1'>residues 73-200</scene> folds with three parallel/antiparallel {{Template:ColorKey_Helix}} packed against six parallel/antiparallel {{Template:ColorKey_Strand}} that form a flat beta-sheet. The B domain has homology with conserved putative <scene name='61/612805/Conserved_g95_and_g164_in_bon/2'>lipid-binding BON</scene> (bacterial OsmY and nodulation) superfamily domains and conserved Gly95 and Gly164 [http://www.ebi.ac.uk/interpro/entry/IPR014004]. <scene name='61/612805/Surface2/1'>The core</scene> is {{Template:ColorKey_Hydrophobic}}, while the exterior is {{Template:ColorKey_Polar}} and predominantly acidic. The two subdomains are symmetric about α2, and their backbone atoms can be aligned with a RMSD [http://http://en.wikipedia.org/wiki/Root-mean-square_deviation] of 1.8Å, by performing a 180° rotation of either one around an axis normal to the 6-stranded β-sheet. | + | The central B domain <scene name='61/612805/Secondary_structure_b/1'>residues 73-200</scene> folds with three parallel/antiparallel {{Template:ColorKey_Helix}} packed against six parallel/antiparallel {{Template:ColorKey_Strand}} that form a flat beta-sheet. |
| | + | <scene name='61/612805/Surface2/1'>The core</scene> is {{Template:ColorKey_Hydrophobic}}, while the exterior is {{Template:ColorKey_Polar}} and predominantly acidic. The two subdomains are symmetric about α2, and their backbone atoms can be aligned with a RMSD [http://http://en.wikipedia.org/wiki/Root-mean-square_deviation] of 1.8Å, by performing a 180° rotation of either one around an axis normal to the 6-stranded β-sheet. |
| | | | |
| | | | |
| Line 26: |
Line 27: |
| | | | |
| | | | |
| - | The C domain <scene name='61/612805/Secondary_structure_c/1'>residues 201-326</scene> has homology to the OmpA-C-like superfamily of periplasmic peptidoglycan-binding sequences, found in several types of bacterial membrane proteins.The C domain of wild-type ArfA <scene name='61/612805/C_domain/3'></scene> <scene name='61/612805/C_domain_1/1'> folds </scene> into four {{Template:ColorKey_Strand}} and four {{Template:ColorKey_Helix}}.Three parallel (β1, β2, β3) and one antiparallel (β4) β-strands form a four-stranded β-sheet (β1–β4-β2–β3) that packs against three α-helices (α1, α2, α3), while a fourth helix (α4) extends from the N-terminus of β4. The structure <scene name='61/612805/C_domain_stabilization/2'> is stabilized </scene> by disulfide bond between C208 and C250, by a network of hydrophobic contacts between α1, α2 and β4 (L211, I215, V243, L247, Ile323 and V325) between side chains and by a hydrogen bond between the backbone amide of V325 and the side-chain carbonyl of Q212. | + | The C domain <scene name='61/612805/Secondary_structure_c/1'>residues 201-326</scene> The C domain of wild-type ArfA <scene name='61/612805/C_domain/3'></scene> <scene name='61/612805/C_domain_1/1'> folds </scene> into four {{Template:ColorKey_Strand}} and four {{Template:ColorKey_Helix}}.Three parallel (β1, β2, β3) and one antiparallel (β4) β-strands form a four-stranded β-sheet (β1–β4-β2–β3) that packs against three α-helices (α1, α2, α3), while a fourth helix (α4) extends from the N-terminus of β4. The structure <scene name='61/612805/C_domain_stabilization/2'> is stabilized </scene> by disulfide bond between C208 and C250, by a network of hydrophobic contacts between α1, α2 and β4 (L211, I215, V243, L247, Ile323 and V325) between side chains and by a hydrogen bond between the backbone amide of V325 and the side-chain carbonyl of Q212. |
| | | | |
| - | ==Putative functions== | + | ==Putative functions== |
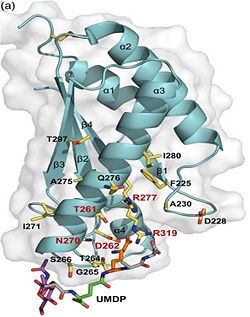
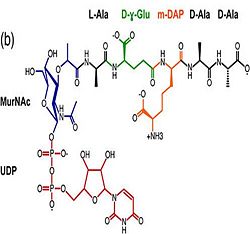
| - | 1. Contribution for structural strength to the bacterial cell wall under acid or other stress conditionsIts functions by binding to peptidoglycan biosyntheesis intermediate uridine-5-'-diphosphate-MurNAc–L-Ala–D-γ-Glu–m-DAP–D-Ala–D-Ala (UMDP) [http://en.wikipedia.org/wiki/Peptidoglycan] binding site. <scene name='61/612805/Peptidoglycan_binding_site/1'>(R277, R319, T261, D262, N270)</scene>. These residues are strictly conserved in the OmpA -like family The side chain of m-DAP is stabilized by charge-charge interactions between its electronegative carbonyl group with R277 and R319 guanidinium groups and with the N270 carboxamide and between its electropositive amino group with the D262 carboxyl and the T261 hydroxyl. UMDP could interact through contacts of its γ-Glu3 and Ala2 backbone amides with the side-chain hydroxyl of S266, of its MurNAc O3 with the amide proton of E267, and of its MurNAc NH2 with the E267 carboxyl <ref>PMID: 22206986 </ref>. | + | 1.The B domain has homology with conserved putative <scene name='61/612805/Conserved_g95_and_g164_in_bon/2'>lipid-binding BON</scene> (bacterial OsmY and nodulation) superfamily domains and conserved Gly95 and Gly164 [http://www.ebi.ac.uk/interpro/entry/IPR014004]. Nitrogen fixation and / or nitrogen metabolism <ref>PMID: 12878000 </ref>. |
| | + | |
| | + | 2. The C domain has homology to the OmpA-C-like superfamily of periplasmic peptidoglycan-binding sequences, found in several types of bacterial membrane proteins.Contribution for structural strength to the bacterial cell wall under acid or other stress conditionsIts functions by binding to peptidoglycan biosyntheesis intermediate uridine-5-'-diphosphate-MurNAc–L-Ala–D-γ-Glu–m-DAP–D-Ala–D-Ala (UMDP) [http://en.wikipedia.org/wiki/Peptidoglycan] binding site. <scene name='61/612805/Peptidoglycan_binding_site/1'>(R277, R319, T261, D262, N270)</scene>. These residues are strictly conserved in the OmpA -like family The side chain of m-DAP is stabilized by charge-charge interactions between its electronegative carbonyl group with R277 and R319 guanidinium groups and with the N270 carboxamide and between its electropositive amino group with the D262 carboxyl and the T261 hydroxyl. UMDP could interact through contacts of its γ-Glu3 and Ala2 backbone amides with the side-chain hydroxyl of S266, of its MurNAc O3 with the amide proton of E267, and of its MurNAc NH2 with the E267 carboxyl <ref>PMID: 22206986 </ref>. |
| | | | |
| | [[Image:123456.jpg|250px]] [[Image:MurNac11.jpg|250px]] | | [[Image:123456.jpg|250px]] [[Image:MurNac11.jpg|250px]] |
| | | | |
| | | | |
| - | 2. Acquisition of Zn(2+) ions by {{Template:ColorKey Composition Ligand}} <scene name='61/612805/Binding-site_for_zn/1'>L82, D96, F97, H125, D127 and V129</scene> <ref>PMID: 22108166 </ref>.
| + | 3. Acquisition of Zn(2+) ions by {{Template:ColorKey Composition Ligand}} <scene name='61/612805/Binding-site_for_zn/1'>binding-site L82, D96, F97, H125, D127 and V129</scene> <ref>PMID: 22108166 </ref>. |
| | | | |
| - | 3. Bacterium's adaptation to the acidic environment (stress response) of the phagosome during infection by:
| + | 4. Bacterium's adaptation to the acidic environment (stress response) of the phagosome during infection by: |
| | a) deamidation of the amino acid pair <scene name='61/612805/Asn111_and_gly112/1'> Asn111-Gly112 </scene>, located at the end of α1 and preceding L3, a pH-dependent reaction whereby Asn is converted to Asp and ammonia is released. Asparagine residues preceding glycine, and situated in conformationally flexible regions of proteins, are frequently deamidated, with potentially significant consequences for protein regulation and function <ref>PMID: 20199110</ref>. [[Image:Asparaginase-reaction.jpg|250px]] | | a) deamidation of the amino acid pair <scene name='61/612805/Asn111_and_gly112/1'> Asn111-Gly112 </scene>, located at the end of α1 and preceding L3, a pH-dependent reaction whereby Asn is converted to Asp and ammonia is released. Asparagine residues preceding glycine, and situated in conformationally flexible regions of proteins, are frequently deamidated, with potentially significant consequences for protein regulation and function <ref>PMID: 20199110</ref>. [[Image:Asparaginase-reaction.jpg|250px]] |
| | | | |
| | b) pH-dependent conformational dynamics of hydrophobic cluster of L232, F225, L240, A244, V281, L285 <scene name='61/612805/D236_before_mutation/1'>in neutral pH (D236) </scene> that folds to a more ordered structure like a flap at <scene name='61/612805/D236a_after_mut/3'>acidic pH (D236A) </scene>. | | b) pH-dependent conformational dynamics of hydrophobic cluster of L232, F225, L240, A244, V281, L285 <scene name='61/612805/D236_before_mutation/1'>in neutral pH (D236) </scene> that folds to a more ordered structure like a flap at <scene name='61/612805/D236a_after_mut/3'>acidic pH (D236A) </scene>. |
| | | | |
| - | 4. Nitrogen fixation and / or nitrogen metabolism <ref>PMID: 12878000 </ref>.
| + | |
| | | | |
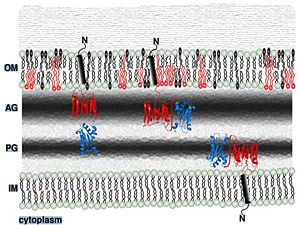
| | ==Putative localization in the outer membrane== | | ==Putative localization in the outer membrane== |
|
Introduction
The Rv0899 ArfA protein [ARFA_MYCTU] from Mycobacterium tuberculosis H37Rv belongs to the OmpA (outer membrane protein A) family of outer membrane proteins and has been proposed to act as an outer membrane porin with ion channel activity and contributes to the bacterium's adaptation to the acidic environment in phagosome [1] during infection [1]. Rv0899 protein encoded by an ammonia release facilator operon that is necessary for rapid ammonia secretion, pH neutralization and adaptation to acidic environments in vitro. Two Mycobacterium tuberculosis H37Rv genes (Rv0900 [ARFB_MYCTU] and Rv0901 [ARFC_MYCTU] ) adjacent to Rv0899 also encode putative membrane proteins, and are found exclusively in association with Rv0899 in the same pathogenic mycobacteria, suggesting that the three may constitute an operon dedicated to a common function.
Asparagine is the primary ammonia source for Mycobacterium tuberculosis H37Rv at acidic pH [2].
The deletion of this gene impairs the uptake of some water-soluble substances, such as serine, glucose and glycerol [3].
Structural highlights
Structure Section
The 326-residue Rv0899 ArfA contains three domains: an N-terminal domain (M domain) (residues 1-72) which includes a sequence of 20 hydrophobic amino acids required for membrane translocation.
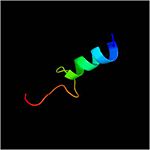
Residues form a mixed alpha/beta-globular structure, encompassing two independently folded modules corresponding to the B and C domains connected by a flexible linker.
The central B domain folds with three parallel/antiparallel Alpha Helices packed against six parallel/antiparallel Beta Strands that form a flat beta-sheet.
is Hydrophobic, while the exterior is Polar and predominantly acidic. The two subdomains are symmetric about α2, and their backbone atoms can be aligned with a RMSD [2] of 1.8Å, by performing a 180° rotation of either one around an axis normal to the 6-stranded β-sheet.
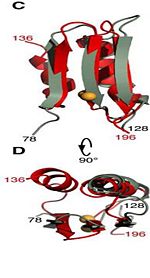
The C domain The C domain of wild-type ArfA into four Beta Strands and four Alpha Helices.Three parallel (β1, β2, β3) and one antiparallel (β4) β-strands form a four-stranded β-sheet (β1–β4-β2–β3) that packs against three α-helices (α1, α2, α3), while a fourth helix (α4) extends from the N-terminus of β4. The structure by disulfide bond between C208 and C250, by a network of hydrophobic contacts between α1, α2 and β4 (L211, I215, V243, L247, Ile323 and V325) between side chains and by a hydrogen bond between the backbone amide of V325 and the side-chain carbonyl of Q212.
Putative functions
1.The B domain has homology with conserved putative (bacterial OsmY and nodulation) superfamily domains and conserved Gly95 and Gly164 [3]. Nitrogen fixation and / or nitrogen metabolism [4].
2. The C domain has homology to the OmpA-C-like superfamily of periplasmic peptidoglycan-binding sequences, found in several types of bacterial membrane proteins.Contribution for structural strength to the bacterial cell wall under acid or other stress conditionsIts functions by binding to peptidoglycan biosyntheesis intermediate uridine-5-'-diphosphate-MurNAc–L-Ala–D-γ-Glu–m-DAP–D-Ala–D-Ala (UMDP) [4] binding site. . These residues are strictly conserved in the OmpA -like family The side chain of m-DAP is stabilized by charge-charge interactions between its electronegative carbonyl group with R277 and R319 guanidinium groups and with the N270 carboxamide and between its electropositive amino group with the D262 carboxyl and the T261 hydroxyl. UMDP could interact through contacts of its γ-Glu3 and Ala2 backbone amides with the side-chain hydroxyl of S266, of its MurNAc O3 with the amide proton of E267, and of its MurNAc NH2 with the E267 carboxyl [5].
3. Acquisition of Zn(2+) ions by Ligand [6].
4. Bacterium's adaptation to the acidic environment (stress response) of the phagosome during infection by:
a) deamidation of the amino acid pair , located at the end of α1 and preceding L3, a pH-dependent reaction whereby Asn is converted to Asp and ammonia is released. Asparagine residues preceding glycine, and situated in conformationally flexible regions of proteins, are frequently deamidated, with potentially significant consequences for protein regulation and function [7]. 
b) pH-dependent conformational dynamics of hydrophobic cluster of L232, F225, L240, A244, V281, L285 that folds to a more ordered structure like a flap at .
Putative localization in the outer membrane
Periplasmic space
References
- ↑ Yang Y, Auguin D, Delbecq S, Dumas E, Molle G, Molle V, Roumestand C, Saint N. Structure of the Mycobacterium tuberculosis OmpATb protein: A model of an oligomeric channel in the mycobacterial cell wall. Proteins. 2010 Oct 12. PMID:21117233 doi:10.1002/prot.22912
- ↑ Song H, Huff J, Janik K, Walter K, Keller C, Ehlers S, Bossmann SH, Niederweis M. Expression of the ompATb operon accelerates ammonia secretion and adaptation of Mycobacterium tuberculosis to acidic environments. Mol Microbiol. 2011 May;80(4):900-18. doi: 10.1111/j.1365-2958.2011.07619.x. Epub, 2011 Mar 16. PMID:21410778 doi:http://dx.doi.org/10.1111/j.1365-2958.2011.07619.x
- ↑ Raynaud C, Papavinasasundaram KG, Speight RA, Springer B, Sander P, Bottger EC, Colston MJ, Draper P. The functions of OmpATb, a pore-forming protein of Mycobacterium tuberculosis. Mol Microbiol. 2002 Oct;46(1):191-201. PMID:12366842
- ↑ Yeats C, Bateman A. The BON domain: a putative membrane-binding domain. Trends Biochem Sci. 2003 Jul;28(7):352-5. PMID:12878000 doi:http://dx.doi.org/10.1016/S0968-0004(03)00115-4
- ↑ Yao Y, Barghava N, Kim J, Niederweis M, Marassi FM. Molecular Structure and Peptidoglycan Recognition of Mycobacterium tuberculosis ArfA (Rv0899). J Mol Biol. 2012 Feb 17;416(2):208-20. Epub 2011 Dec 21. PMID:22206986 doi:10.1016/j.jmb.2011.12.030
- ↑ Li J, Shi C, Gao Y, Wu K, Shi P, Lai C, Chen L, Wu F, Tian C. Structural Studies of Mycobacterium tuberculosis Rv0899 Reveal a Monomeric Membrane-Anchoring Protein with Two Separate Domains. J Mol Biol. 2011 Nov 15. PMID:22108166 doi:10.1016/j.jmb.2011.11.016
- ↑ Teriete P, Yao Y, Kolodzik A, Yu J, Song H, Niederweis M, Marassi FM. Mycobacterium tuberculosis Rv0899 Adopts a Mixed alpha/beta-Structure and Does Not Form a Transmembrane beta-Barrel. Biochemistry. 2010 Mar 10. PMID:20199110 doi:10.1021/bi100158s
|






