This old version of Proteopedia is provided for student assignments while the new version is undergoing repairs. Content and edits done in this old version of Proteopedia after March 1, 2026 will eventually be lost when it is retired in about June of 2026.
Apply for new accounts at the new Proteopedia. Your logins will work in both the old and new versions.
Connexin
From Proteopedia
| Line 1: | Line 1: | ||
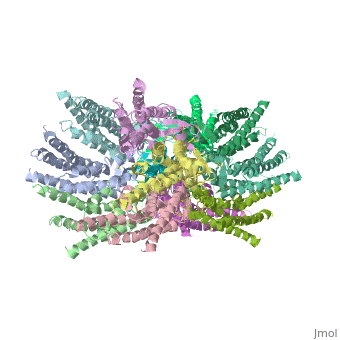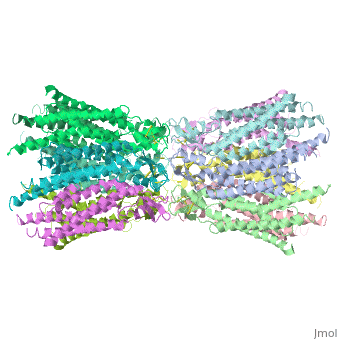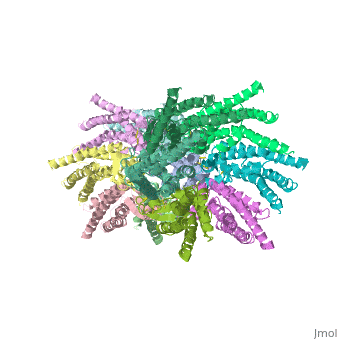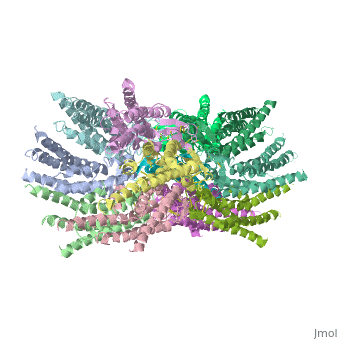
| - | == | + | ==Connexins are integral transmembrane proteins that form intercellular channels in vertebrates. Six connexins form a hexamerical assembly, known as connexon or hemichannel, which delineates an aqueous pore with a minimum diameter of ∼1.2 nm. When two hemichannels from adjacent cells dock and join, leaving a gap of ∼2–3 nm, they may form an intercellular gap junction channel [[http://www.uniprot.org/uniprot/P29033]] which spans the two plasma membranes and allows the exchange of cytoplasmic molecules with size up to ∼1 kDa. The importance of electrical and molecular signaling through gap junction channels is widely recognized . Virtually all cells in solid tissues are coupled by gap junctions , thus it is not surprising that mutations in connexin genes have been linked to a variety of human diseases, including cardiovascular anomalies, peripheral neuropathy, skin disorders, cataracts, and deafness. Of notice, about half of all cases of human deafness in countries surrounding the Mediterranean have been linked to mutations in the GJB2 gene [[http://www.uniprot.org/uniprot/P29033]], which encodes Cx26 .<ref name='important'>pmid 24624091</ref>. |
| + | GJB2 is a gene which encodes a member of the gap junction protein family. Intercellular signaling is one of the most essential properties of multicellular organisms. Gap junctions [[http://www.uniprot.org/uniprot/P29033]] are specialized membrane regions containing hundreds of intercellular communication channels that allow the passage of molecules such as ions, metabolites, nucleotides and small peptides. The gap junctions were first characterized by electron microscopy as regionally specialized structures on plasma membranes of contacting adherent cells. These structures were shown to consist of cell-to-cell channels that facilitate the transfer of ions and small molecules between cells. The gap junction proteins, also known as connexins, purified from fractions of enriched gap junctions from different tissues differ. The gap junction proteins are divided into two categories, alpha and beta. Mutations in this gene are responsible for as much as 50% of pre-lingual, recessive deafness. <ref name='Structure'>pmid 19622859</ref> | ||
| + | |||
<StructureSection load='2ZW3' size='340' side='right' caption='Caption for this structure' scene=''> | <StructureSection load='2ZW3' size='340' side='right' caption='Caption for this structure' scene=''> | ||
| - | '''''Introduction:''''' | ||
| - | Connexins are integral transmembrane proteins that form intercellular channels in vertebrates. Six connexins form a hexamerical assembly, known as connexon or hemichannel, which delineates an aqueous pore with a minimum diameter of ∼1.2 nm. When two hemichannels from adjacent cells dock and join, leaving a gap of ∼2–3 nm, they may form an intercellular gap junction channel [[http://www.uniprot.org/uniprot/P29033]] which spans the two plasma membranes and allows the exchange of cytoplasmic molecules with size up to ∼1 kDa. The importance of electrical and molecular signaling through gap junction channels is widely recognized . Virtually all cells in solid tissues are coupled by gap junctions , thus it is not surprising that mutations in connexin genes have been linked to a variety of human diseases, including cardiovascular anomalies, peripheral neuropathy, skin disorders, cataracts, and deafness. Of notice, about half of all cases of human deafness in countries surrounding the Mediterranean have been linked to mutations in the GJB2 gene [[http://www.uniprot.org/uniprot/P29033]], which encodes Cx26 .<ref name='important'>pmid 24624091</ref>. | ||
| - | GJB2 is a gene which encodes a member of the gap junction protein family. Intercellular signaling is one of the most essential properties of multicellular organisms. Gap junctions [[http://www.uniprot.org/uniprot/P29033]] are specialized membrane regions containing hundreds of intercellular communication channels that allow the passage of molecules such as ions, metabolites, nucleotides and small peptides. The gap junctions were first characterized by electron microscopy as regionally specialized structures on plasma membranes of contacting adherent cells. These structures were shown to consist of cell-to-cell channels that facilitate the transfer of ions and small molecules between cells. The gap junction proteins, also known as connexins, purified from fractions of enriched gap junctions from different tissues differ. The gap junction proteins are divided into two categories, alpha and beta. Mutations in this gene are responsible for as much as 50% of pre-lingual, recessive deafness. <ref name='Structure'>pmid 19622859</ref> | ||
| - | <scene name='70/701426/Connexin_26_basic_structure/1'>TextToBeDisplayed</scene> | ||
| - | ''''' | + | '''''Phenotypes of mutations in connexin 26?''''' |
| - | '' | + | |
| - | ''' | + | |
Mutations in human Connexin26 (hCx26) can lead to congenital hearing loss (1 child per 1000 frequency) that can be syndromic or non-syndromic. Non-syndromic hearing loss (NSHL) is characterized by sensorineural hearing loss in the absence of other symptoms, while syndromic hearing loss affects other organ systems, primarily the skin. mutations in GJB2 (the gene that encodes for Cx26) account for about half of all congenital and autosomal recessive nonsyndromic hearing loss in every population tested . Although the most frequently occurring NSHL mutations produce severely truncated proteins due to frameshift or missense, almost 80% of the known deafness mutations are actually single amino acid changes or deletions. These mutations have been found across the entire sequence of Cx26. The majority of NSHL mutations cause either generalized folding problems that result in the failure of Cx26 to traffic to the cell surface, or are permissive for the formation of gap junction plaques, but prevent intercellular channel function.<ref name='mutant int'>pmid 23967136</ref> | Mutations in human Connexin26 (hCx26) can lead to congenital hearing loss (1 child per 1000 frequency) that can be syndromic or non-syndromic. Non-syndromic hearing loss (NSHL) is characterized by sensorineural hearing loss in the absence of other symptoms, while syndromic hearing loss affects other organ systems, primarily the skin. mutations in GJB2 (the gene that encodes for Cx26) account for about half of all congenital and autosomal recessive nonsyndromic hearing loss in every population tested . Although the most frequently occurring NSHL mutations produce severely truncated proteins due to frameshift or missense, almost 80% of the known deafness mutations are actually single amino acid changes or deletions. These mutations have been found across the entire sequence of Cx26. The majority of NSHL mutations cause either generalized folding problems that result in the failure of Cx26 to traffic to the cell surface, or are permissive for the formation of gap junction plaques, but prevent intercellular channel function.<ref name='mutant int'>pmid 23967136</ref> | ||
Connexin26 (CX26) protein is essential for maintaining the high K+ concentration in the endolymph of the inner ear. Sound stimulation of the ossicular chain causes vibrations in the endolymph .K+ ions enter the hair cells under the influence of these vibrations and vibration signal is ultimately converted into a neural signal. The system is regenerated by the release of K+ from the hair cells into the supporting cells. The K+ ions are then passed from cell to cell via gap junctions and are eventually released into the endolymph. Except for sensorineural cells, the CX26 protein is present in gap junctions connecting all cell types in the cochlea , including the spiral limbus, the supporting cells, the spiral ligament and the basal and the basal and intermediate cells of the stria vascularis. It is therefore very likely that connexin 26 is involved in K+ -recycling in the cochlea. | Connexin26 (CX26) protein is essential for maintaining the high K+ concentration in the endolymph of the inner ear. Sound stimulation of the ossicular chain causes vibrations in the endolymph .K+ ions enter the hair cells under the influence of these vibrations and vibration signal is ultimately converted into a neural signal. The system is regenerated by the release of K+ from the hair cells into the supporting cells. The K+ ions are then passed from cell to cell via gap junctions and are eventually released into the endolymph. Except for sensorineural cells, the CX26 protein is present in gap junctions connecting all cell types in the cochlea , including the spiral limbus, the supporting cells, the spiral ligament and the basal and the basal and intermediate cells of the stria vascularis. It is therefore very likely that connexin 26 is involved in K+ -recycling in the cochlea. | ||
| Line 28: | Line 24: | ||
The short NTHs of the six protomers formthe funnel , This finding agrees with an NMR solution structure of an N-terminal peptide of Cx26, which showed that the loop connecting the NTH to TM1 is very flexible30. Asp 2 forms hydrogen bonds with the mainchain amide of Thr 5 from the neighbouring protomer. The Asp 2 and Thr 5 residues on neighbouring NTHs at the bottom of the funnel form a circular girdle, as previously seen in the nicotinic acetylcholine receptor31, which stabilizes the funnel structure . <ref name='Structure'/> | The short NTHs of the six protomers formthe funnel , This finding agrees with an NMR solution structure of an N-terminal peptide of Cx26, which showed that the loop connecting the NTH to TM1 is very flexible30. Asp 2 forms hydrogen bonds with the mainchain amide of Thr 5 from the neighbouring protomer. The Asp 2 and Thr 5 residues on neighbouring NTHs at the bottom of the funnel form a circular girdle, as previously seen in the nicotinic acetylcholine receptor31, which stabilizes the funnel structure . <ref name='Structure'/> | ||
| - | '''''Differences between wild type and | + | '''''Differences between wild type and mutant connexin 26 :''''' |
In general, single site mutations are spread fairly evenly across the whole protein with TM2 having the highest mutation density (number of amino acids with NHLS mutations divided by the total number of amino acids in the domain) at 67% to M1 and E1 having the lowest density of mutations with their respective domains at 33%. According to this criterion, TM4 has a mutation density of 40%. . Of the four transmembrane helices, M1, M2 and M3 have attracted the most attention, because of the controversies involved in models with different helix assignments, based on lower resolution cryo-electron crystallographic structures and scanning cysteine accessibility mutagenesis . Far less is known about TM4 and how side chains interact with the other helices and with the lipid bilayer. <ref name='mutant int'/> | In general, single site mutations are spread fairly evenly across the whole protein with TM2 having the highest mutation density (number of amino acids with NHLS mutations divided by the total number of amino acids in the domain) at 67% to M1 and E1 having the lowest density of mutations with their respective domains at 33%. According to this criterion, TM4 has a mutation density of 40%. . Of the four transmembrane helices, M1, M2 and M3 have attracted the most attention, because of the controversies involved in models with different helix assignments, based on lower resolution cryo-electron crystallographic structures and scanning cysteine accessibility mutagenesis . Far less is known about TM4 and how side chains interact with the other helices and with the lipid bilayer. <ref name='mutant int'/> | ||
Revision as of 09:22, 13 May 2015
==Connexins are integral transmembrane proteins that form intercellular channels in vertebrates. Six connexins form a hexamerical assembly, known as connexon or hemichannel, which delineates an aqueous pore with a minimum diameter of ∼1.2 nm. When two hemichannels from adjacent cells dock and join, leaving a gap of ∼2–3 nm, they may form an intercellular gap junction channel [[1]] which spans the two plasma membranes and allows the exchange of cytoplasmic molecules with size up to ∼1 kDa. The importance of electrical and molecular signaling through gap junction channels is widely recognized . Virtually all cells in solid tissues are coupled by gap junctions , thus it is not surprising that mutations in connexin genes have been linked to a variety of human diseases, including cardiovascular anomalies, peripheral neuropathy, skin disorders, cataracts, and deafness. Of notice, about half of all cases of human deafness in countries surrounding the Mediterranean have been linked to mutations in the GJB2 gene [[2]], which encodes Cx26 .[1]. GJB2 is a gene which encodes a member of the gap junction protein family. Intercellular signaling is one of the most essential properties of multicellular organisms. Gap junctions [[3]] are specialized membrane regions containing hundreds of intercellular communication channels that allow the passage of molecules such as ions, metabolites, nucleotides and small peptides. The gap junctions were first characterized by electron microscopy as regionally specialized structures on plasma membranes of contacting adherent cells. These structures were shown to consist of cell-to-cell channels that facilitate the transfer of ions and small molecules between cells. The gap junction proteins, also known as connexins, purified from fractions of enriched gap junctions from different tissues differ. The gap junction proteins are divided into two categories, alpha and beta. Mutations in this gene are responsible for as much as 50% of pre-lingual, recessive deafness. [2]
| |||||||||||

References
- ↑ Zonta F, Buratto D, Cassini C, Bortolozzi M, Mammano F. Molecular dynamics simulations highlight structural and functional alterations in deafness-related M34T mutation of connexin 26. Front Physiol. 2014 Mar 4;5:85. doi: 10.3389/fphys.2014.00085. eCollection 2014. PMID:24624091 doi:http://dx.doi.org/10.3389/fphys.2014.00085
- ↑ 2.0 2.1 2.2 2.3 2.4 Suga M, Maeda S, Nakagawa S, Yamashita E, Tsukihara T. A description of the structural determination procedures of a gap junction channel at 3.5 A resolution. Acta Crystallogr D Biol Crystallogr. 2009 Aug;65(Pt 8):758-66. Epub 2009, Jul 10. PMID:19622859 doi:http://dx.doi.org/10.1107/S0907444909014711
- ↑ 3.0 3.1 Ambrosi C, Walker AE, Depriest AD, Cone AC, Lu C, Badger J, Skerrett IM, Sosinsky GE. Analysis of trafficking, stability and function of human connexin 26 gap junction channels with deafness-causing mutations in the fourth transmembrane helix. PLoS One. 2013 Aug 15;8(8):e70916. doi: 10.1371/journal.pone.0070916. eCollection, 2013. PMID:23967136 doi:http://dx.doi.org/10.1371/journal.pone.0070916
- ↑ 4.0 4.1 Oshima A, Tani K, Toloue MM, Hiroaki Y, Smock A, Inukai S, Cone A, Nicholson BJ, Sosinsky GE, Fujiyoshi Y. Asymmetric Configurations and N-terminal Rearrangements in Connexin26 Gap Junction Channels. J Mol Biol. 2011 Jan 21;405(3):724-35. Epub 2010 Nov 20. PMID:21094651 doi:10.1016/j.jmb.2010.10.032
Proteopedia Page Contributors and Editors (what is this?)
Safaa Salah Hussiesy, Michal Harel, Doaa Naffaa, Jaime Prilusky

{kind=link}