This old version of Proteopedia is provided for student assignments while the new version is undergoing repairs. Content and edits done in this old version of Proteopedia after March 1, 2026 will eventually be lost when it is retired in about June of 2026.
Apply for new accounts at the new Proteopedia. Your logins will work in both the old and new versions.
Connexin
From Proteopedia
(Difference between revisions)
| Line 1: | Line 1: | ||
<StructureSection load='2ZW3' size='340' side='right' caption='Caption for this structure' scene='> | <StructureSection load='2ZW3' size='340' side='right' caption='Caption for this structure' scene='> | ||
=Introduction= | =Introduction= | ||
| - | [http://en.wikipedia.org/wiki/Connexin Connexins] are integral transmembrane [http://en.wikipedia.org/wiki/Protein proteins] that form intercellular channels in [http://en.wikipedia.org/wiki/Vertebrate vertebrates]. Connexin 26 is encoded by the [http://www.uniprot.org/uniprot/P29033 GJB2 gene]. Six connexins form a hexamerical assembly, known as [http://en.wikipedia.org/wiki/Connexon connexon] or hemichannel, which form an intercellular [http://en.wikipedia.org/wiki/Gap_junction gap junction channel]. Gap junctions are specialized membrane regions containing hundreds of intercellular communication channels that allow the passage of molecules such as ions, metabolites, nucleotides and small peptides. Virtually all cells in solid tissues are coupled by gap junctions, thus it is not surprising that mutations in connexin genes have been linked to a variety of [http://omim.org/entry/121011?search=gjb2%20deafness-causing&highlight=deafnesscausing%20deafness%20gjb2%20deaf%20causing Connexin human diseases], including cardiovascular anomalies, peripheral neuropathy, skin disorders, [http://en.wikipedia.org/wiki/Cataract cataracts], and deafness. | + | [http://en.wikipedia.org/wiki/Connexin Connexins] are integral transmembrane [http://en.wikipedia.org/wiki/Protein proteins] that form intercellular channels in [http://en.wikipedia.org/wiki/Vertebrate vertebrates]. Connexin 26 is encoded by the [http://www.uniprot.org/uniprot/P29033 GJB2 gene]. Six connexins form a hexamerical assembly, known as [http://en.wikipedia.org/wiki/Connexon a connexon] or a hemichannel, which form an intercellular [http://en.wikipedia.org/wiki/Gap_junction gap junction channel]. Gap junctions are specialized membrane regions containing hundreds of intercellular communication channels that allow the passage of molecules such as ions, metabolites, nucleotides and small peptides. Virtually all cells in solid tissues are coupled by gap junctions, thus it is not surprising that mutations in connexin genes have been linked to a variety of [http://omim.org/entry/121011?search=gjb2%20deafness-causing&highlight=deafnesscausing%20deafness%20gjb2%20deaf%20causing Connexin human diseases], including cardiovascular anomalies, peripheral neuropathy, skin disorders, [http://en.wikipedia.org/wiki/Cataract cataracts], and deafness. |
Of notice, about half of all cases of human pre-lingual recessive deafness in countries surrounding the Mediterranean have been linked to mutations in the GJB2 gene<ref name='important'>pmid 24624091</ref>,<ref name='Structure'>pmid 19622859</ref> | Of notice, about half of all cases of human pre-lingual recessive deafness in countries surrounding the Mediterranean have been linked to mutations in the GJB2 gene<ref name='important'>pmid 24624091</ref>,<ref name='Structure'>pmid 19622859</ref> | ||
| Line 8: | Line 8: | ||
| - | + | ==Phenotypic results of mutations in connexin 26 and hearing loss== | |
| - | + | ||
| - | + | ||
| - | ==Phenotypic results of mutations in connexin 26== | + | |
Mutations in human Connexin 26 (hCx26) can lead to [http://en.wikipedia.org/wiki/Congenital_hearing_loss congenital hearing loss] <ref>pmid 25153233</ref> (1 child per 1000 frequency) that can be syndromic or non-syndromic. Non-syndromic hearing loss (NSHL) is characterized by sensorineural hearing loss in the absence of other symptoms, while syndromic hearing loss affects other organ systems, primarily the skin. mutations in GJB2 (the gene that encodes for Cx26) account for about half of all congenital and autosomal recessive nonsyndromic hearing loss in every population tested . Although the most frequently occurring (NSHL) mutations produce severely truncated proteins due to frameshift or missense, almost 80% of the known deafness mutations are actually single amino acid changes or deletions. These mutations have been found across the entire sequence of Cx26. The majority of NSHL mutations cause either generalized folding problems that result in the failure of Cx26 to traffic to the cell surface, or are permissive for the formation of gap junction plaques, but prevent intercellular channel function.<ref name='mutant int'>pmid 23967136</ref> | Mutations in human Connexin 26 (hCx26) can lead to [http://en.wikipedia.org/wiki/Congenital_hearing_loss congenital hearing loss] <ref>pmid 25153233</ref> (1 child per 1000 frequency) that can be syndromic or non-syndromic. Non-syndromic hearing loss (NSHL) is characterized by sensorineural hearing loss in the absence of other symptoms, while syndromic hearing loss affects other organ systems, primarily the skin. mutations in GJB2 (the gene that encodes for Cx26) account for about half of all congenital and autosomal recessive nonsyndromic hearing loss in every population tested . Although the most frequently occurring (NSHL) mutations produce severely truncated proteins due to frameshift or missense, almost 80% of the known deafness mutations are actually single amino acid changes or deletions. These mutations have been found across the entire sequence of Cx26. The majority of NSHL mutations cause either generalized folding problems that result in the failure of Cx26 to traffic to the cell surface, or are permissive for the formation of gap junction plaques, but prevent intercellular channel function.<ref name='mutant int'>pmid 23967136</ref> | ||
| + | |||
| + | ==The biochemical function of connexin 26 in the ear== | ||
| + | Connexin 26 (CX26) protein is essential for maintaining the high K+ concentration in the [http://en.wikipedia.org/wiki/Endolymph endolymph] of the inner ear. Sound stimulation of the ossicular chain causes vibrations in the [http://en.wikipedia.org/wiki/Endolymph endolymph]. K+ ions enter the hair cells under the influence of these vibrations and vibration signal is ultimately converted into a neural signal. The system is regenerated by the release of K+ from the hair cells into the supporting cells. The K+ ions are then passed from cell to cell via gap junctions and are eventually released into the endolymph, Except for sensorineural cells, the CX26 protein is present in gap junctions connecting all cell types in the [http://en.wikipedia.org/wiki/Cochlea cochlea], including the spiral limbus, the supporting cells, the spiral ligament and the basal and intermediate cells of the [http://en.wikipedia.org/wiki/Stria_vascularis_of_cochlear_duct stria vascularis]. It is therefore very likely that connexin 26 is involved in K+ recycling in the [http://en.wikipedia.org/wiki/Cochlea cochlea].<ref name='function'>pmid 11810458</ref> | ||
| Line 29: | Line 29: | ||
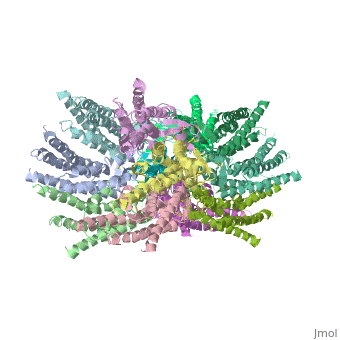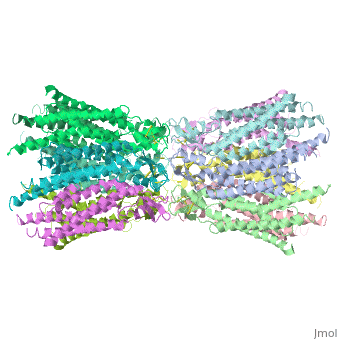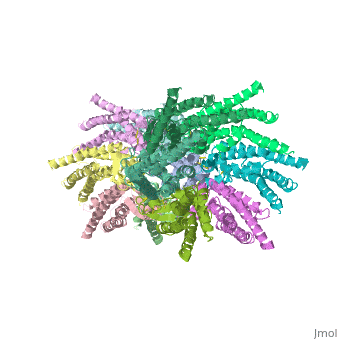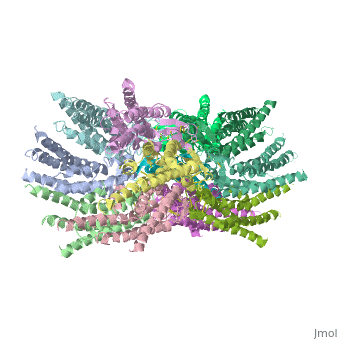
==Structure of the cx26 protomer:== | ==Structure of the cx26 protomer:== | ||
| - | As mentioned before <scene name='70/701426/Connexin_structure/1'>the connexin</scene> [http://en.wikipedia.org/wiki/Promoter_(genetics) protomer] has four transmembrane (TM1–4), two extracellular loops , a cytoplasmic loop, an N-terminal helix (NTH), and a C-terminal segment. Cx26 forms a typical four-helix bundle in which any pair of adjacent helices is antiparallel. The major pore-lining helix TM1 is inclined, so that the pore diameter narrows from the cytoplasmic to the extracellular side of the membrane, and ends in a short [http://en.wikipedia.org/wiki/310_helix 3<sub>10</sub> helix]. | + | As mentioned before <scene name='70/701426/Connexin_structure/1'>the connexin</scene> [http://en.wikipedia.org/wiki/Promoter_(genetics) protomer] has four transmembrane (TM1–4), two extracellular loops, a cytoplasmic loop, an N-terminal helix (NTH), and a C-terminal segment. Cx26 forms a typical four-helix bundle in which any pair of adjacent helices is antiparallel. The major pore-lining helix TM1 is inclined, so that the pore diameter narrows from the cytoplasmic to the extracellular side of the membrane, and ends in a short [http://en.wikipedia.org/wiki/310_helix 3<sub>10</sub> helix]. |
The extracellular loop E1 contains a [http://en.wikipedia.org/wiki/310_helix 3<sub>10</sub> helix] at the beginning and a short α-helix in its C-terminal. E2, together with E1, contains a short antiparallel β-sheet and stretches over E1, forming the outside <scene name='70/701426/Connexon_bachbone/1'>wall</scene> of the connexon. Six conserved cysteine residues, three in each loop, form intramolecular disulphide bonds between E1 and E2 Most of the prominent intra-protomer interactions are in the extracellular part of the transmembrane region, The interactions between the two adjoining connexons of the gap junction channel, which involve both E1 and E2. [[image:E1.jpg | thumb | center | 250px | Disulfide bonds between two extracellular loops in the Cx26 promoter]] | The extracellular loop E1 contains a [http://en.wikipedia.org/wiki/310_helix 3<sub>10</sub> helix] at the beginning and a short α-helix in its C-terminal. E2, together with E1, contains a short antiparallel β-sheet and stretches over E1, forming the outside <scene name='70/701426/Connexon_bachbone/1'>wall</scene> of the connexon. Six conserved cysteine residues, three in each loop, form intramolecular disulphide bonds between E1 and E2 Most of the prominent intra-protomer interactions are in the extracellular part of the transmembrane region, The interactions between the two adjoining connexons of the gap junction channel, which involve both E1 and E2. [[image:E1.jpg | thumb | center | 250px | Disulfide bonds between two extracellular loops in the Cx26 promoter]] | ||
| - | The N-terminal half of E2 seems rather flexible and its amino-acid sequence varies greatly among connexins . The C-terminal half of E2 begins with a | + | The N-terminal half of E2 seems rather flexible and its amino-acid sequence varies greatly among connexins. The C-terminal half of E2 begins with a 3<sub>10</sub> turn is followed by a conserved Pro-Cys-Pro motif that reverses its direction back to TM4. Most of the prominent intra-protomer interactions are in the extracellular part of the transmembrane region. Arg 32 (TM1) interactswithGln 80 (TM2), Glu 147 (TM3) and Ser 199 (TM4). Two hydrophobic cores around Trp 44 (E1) and Trp 77 (TM2) stabilize the protomer structure. Ala 39 (TM1), Ala 40 (TM1), Val 43 (E1) and Ile 74 (TM2) contribute to the first hydrophobic core around Trp 44, and Phe 154 (TM3) and Met 195 (TM4) form the second core with Trp 77. In the intracellular part of the transmembrane region, Arg 143 (TM3) forms hydrogen bonds with Asn 206 (TM3) and Ser 139 (TM3).<ref name='Structure'/> |
[[image:bonds.jpg | thumb | center | 500px | Intracellular interactions]] | [[image:bonds.jpg | thumb | center | 500px | Intracellular interactions]] | ||
=Differences between wild type and mutant connexin 26:= | =Differences between wild type and mutant connexin 26:= | ||
| - | In general, single site mutations are spread fairly evenly across the whole protein with TM2 having the highest mutation density (number of amino acids with NHLS mutations divided by the total number of amino acids in the domain) at 67% to M1 and E1, having the lowest density of mutations with their respective domains at 33%. According to this criterion, TM4 has a mutation density of 40%. Of the four transmembrane helices, M1, M2 and M3 have attracted the most attention, because of the controversies involved in models with different helix assignments, based on lower resolution cryo-electron crystallographic structures and scanning cysteine accessibility mutagenesis. Far less is known about TM4 and how side chains interact with the other helices and with the lipid bilayer. <ref name='mutant int'/> | + | In general, single site mutations are spread fairly evenly across the whole protein with TM2 having the highest mutation density (number of amino acids with NHLS mutations divided by the total number of amino acids in the domain) at 67% to M1 and E1, having the lowest density of mutations with their respective domains at 33%. According to this criterion, TM4 has a mutation density of 40%. Of the four transmembrane helices, M1, M2 and M3 have attracted the most attention, because of the controversies involved in models with different helix assignments, based on lower resolution cryo-electron crystallographic structures and scanning cysteine accessibility mutagenesis. Far less is known about TM4 and how side chains interact with the other helices and with the lipid bilayer.<ref name='mutant int'/> |
Two structural crystallographic studies have been commenced on Cx26, the first one describing the WT protein in a resolution of 3.5 Å by Tsuhikara, T.(2009)<ref name='Structure'/>, and the second one deals with two types of mutations in the N terminus of the protein by Fujiyoshi, Y.(2011).<ref name='pdb'/> | Two structural crystallographic studies have been commenced on Cx26, the first one describing the WT protein in a resolution of 3.5 Å by Tsuhikara, T.(2009)<ref name='Structure'/>, and the second one deals with two types of mutations in the N terminus of the protein by Fujiyoshi, Y.(2011).<ref name='pdb'/> | ||
Revision as of 07:28, 11 August 2015
| |||||||||||




Proteopedia Page Contributors and Editors (what is this?)
Safaa Salah Hussiesy, Michal Harel, Doaa Naffaa, Jaime Prilusky
