Function
Human growth hormone (hGH) plays a vital role in growth and development. It is naturally produced by somatotropic cells in the anterior pituitary gland. The hormone is produced as a 217 amino acid precursor protein. The 26 N-terminal amino acids correspond to a signal peptide, which is essential for hormone secretion. This signal peptide is cleaved during the secretion process to yield the mature, 191 amino acid form of hGH.
Mature hGH travels through the bloodstream and interacts with a specific hGH-receptor on the surface of various cells, including muscle, bone, and cartilage. Binding of hGH to its receptor causes dimerization and signal transduction, which ultimately stimulates cellular division. HGH also indirectly influences growth by stimulating the liver to produce additional growth factors, such as insulin-like growth factor-1. Synthetic versions of hGH produced by recombinant DNA technology are used to treat growth disorders associated with hGH deficiencies. Prolactin receptor (PRLR) can also bind to and be activated by growth hormone.
Location in the Body
hGH is produced in the anterior pituitary gland or hypophyse and is located at the base of the brain. The hormones then circulate throughout the vascular system of the body. The pituitary gland helps control growth as well as blood pressure, energy management, all functions of the sex organs, thyroid glands and metabolism, temperature regulation and pain relief. hGH interacts with a variety of cells while in the bloodstream including muscle and bone cells. It eventually reaches and stimulates growth of all major organs (particularly the liver where it stimulates the production of growth factors) with the exception of the brain.
Growth Hormone’s Signal Peptide
Human growth hormone exists in two forms, pre-GH and mature GH. The pre-protein contains a 26 residue-long signal peptide located on the N-terminus of the protein, which is cleaved off to create the mature protein.
The signal peptide acts as an anchor to keep the pre-protein bound to the membrane of the endoplasmic reticulum (ER). Following proper signaling, an ER membrane-bound signal I peptidase cleaves the 217 amino acid long pre-protein between residues 26 and 27 resulting in the 191 residue-long mature secretory GH (Chawla 1983; Tuteja 2005)[1]. The now active protein releases from the membrane into the ER lumen and is exported out of the cell.
Mutations to residues within the signal peptide have show to affect the secretion of GH from the cell. Specifically, the Lys16Pro variant is associated with decreased levels of GH due to suppressed secretion, resulting in growth hormone deficiency isolated type 1B (IGHD1B) (Millar D.S. 2003)[2].
Isoforms
There are four natural splice variants, or isoforms, of human growth hormone that have been identified. The different isoforms are produced when splicing of the five exons occurs at various positions. Note that in all four isoforms, the first 26 amino acids on the N-terminus are only present in the precursor form, and are not part of the active hormone. The 22KD GH (22 K-GH) is the most prominently found variant and is deemed as the canonical sequence, meaning that it serves as the reference to the other isoforms in terms of location of features. Is is followed by 20KD GH(20 K-GH) and other rare isoforms. One of the key differences about the other isoforms is that since they are lacking certain sections of amino acids, then the single point mutations that naturally occur in those regions will not affect the function or activity of the isoform proteins. For example, there several single point mutations that occur in the range of amino acids 58 through 72 that result in reduced ability to activate the JAK/STAT pathway (Millar et al., 2003). This section is absent in isoform 2, and therefore that variant of the protein is not susceptible to such alterations. Similarly, there are single point mutations in the range of amino acids 111 through 148 and 117 through 162 that result in reduced secretion (Millar et al., 2003), reduced ability to activate the JAK/STAT pathway (Millar et al., 2003), and loss of activity (Takahashi et al., 1997)[3]. Therefore, since isoforms 2 and 3 are lacking these regions, they are not subject to these various effects on activity.
Isoform 1 (GH1) (191 amino acids; MW = 24,847 kDa)
The GH1 is expressed mainly in somatotrope cells of the pituitary gland. This 22 K-GH molecule is the main GH isoform, representing more than 90% of total GH in circulation. Its tertiary structure is a 4-helical twisted bundle with unusual connectivity. The helices run up-up-down-down instead of the more usual up-down-up-down form.
GH is best known from its growth promoting activity in children, but also has import- ant biological activities in adults. These include lipolysis, glucose-, calcium- and phosphorous-metabolism as well as lactogenesis and immune function.
MATGSRTSLL LAFGLLCLPW LQEGSAFPTI PLSRLFDNAM LRAHRLHQLA FDTYQEFEEAYIPKEQKYSF LQNPQTSLCF SESIPTPSNR EETQQKSNLE LLRISLLLIQ SWLEPVQFLR SVFANSLVYG ASDSNVYDLL KDLEEGIQTL MGRLEDGSPR TGQIFKQTYS KFDTNSHNDDALLKNYGLLY CFRKDMDKVE TFLRIVQCRS VEGSCGF
Isoform 2 (176 amino acids; MW = 22,992 kDa; Missing amino acids 58-72)
The second most abundant GH isoform is the 20 K-GH molecule. It is derived from GH-1 by alternative pre-messenger ribonucleic acid (pre mRNA) splicing of exon 3. The structure is similar to 22 K-GH except for a de- letion of the internal residues 32–46. Therefore, 20 K-GH consists of 176 amino acids only.
There is compelling evidences that both 22 K- and 20 K-GH can activate Janus Kinase 2 (JAK2), signal transducers and activators of tran- scription 1, 3 and 5 (STATs 1/3/5), although the level of STAT 1/3/5 phosphorylation induced by 22 K-GH are higher than those of 20 K-GH [18].
MATGSRTSLL LAFGLLCLPW LQEGSAFPTI PLSRLFDNAM LRAHRLHQLA FDTYQEFNPQTSLCFSESIP TPSNREETQQ KSNLELLRIS LLLIQSWLEP VQFLRSVFAN SLVYGASDSNVYDLLKDLEE GIQTLMGRLE DGSPRTGQIF KQTYSKFDTN SHNDDALLKN YGLLYCFRKDMDKVETFLRI VQCRSVEGSC GF
Isoform 3 (153 amino acids; MW = 20,561 kDa; Missing amino acids 111-148)
MATGSRTSLL LAFGLLCLPW LQEGSAFPTI PLSRLFDNAM LRAHRLHQLA FDTYQEFEEAYIPKEQKYSF LQNPQTSLCF SESIPTPSNR EETQQKSNLE LLRISLLLIQ TLMGRLEDGSPRTGQIFKQT YSKFDTNSHN DDALLKNYGL LYCFRKDMDK VETFLRIVQC RSVEGSCGF
Isoform 4 (145 amino acids; MW = 19,802 kDa; Missing amino acids 117-162)
MATGSRTSLL LAFGLLCLPW LQEGSAFPTI PLSRLFDNAM LRAHRLHQLA FDTYQEFEEAYIPKEQKYSF LQNPQTSLCF SESIPTPSNR EETQQKSNLE LLRISLLLIQ SWLEPVQIFKQTYSKFDTNS HNDDALLKNY GLLYCFRKDM DKVETFLRIV QCRSVEGSCG F
Structure
Primary Sequence
hGH is produced within the cell as a pre-protein, consisting of a 191 polypeptide chain associated with a 26 amino acid signal peptide. This signal peptide functions in membrane translocation, and is eventually cleaved to yield the mature form of hGH-1. The signal peptide is cleaved from the 191 polypeptide chain of hGH-1 by an ER membrane-bound protease (Chawla, Parks, and Rudman, 1983)
MATGSRTSLLLAFGLLCLPWLQEGSAFPTIPLSRLFDNAMLRAHRLHQLAFDTYQEF
EAYIPKEQKYSFLQNPQTSLCFSESIPTPSNREETQQKSNLELLRISLLLIQSWLEPVQFR
SVFANSLVYGASDSNVYDLLKDLEEGIQTLMGRLEDGSPRTGQIFKQTYSKFDTNSH
NDALLKNYGLLYCFRKDMDKVETFLRIVQCRSVEGSCGF
Primary Sequence of hGH-1 precursor
Primary Sequence Variations between Isoforms:
hGH-1 (most abundant isoform): has a primary amino acid sequence of 191 amino acids
Isoform 2: amino acid residues 58-72 of hGH-1 are excluded
Isoform 3: excludes amino acid residues 111-148
Isoform 4: excludes amino acid residues 117-162
Secondary Structure
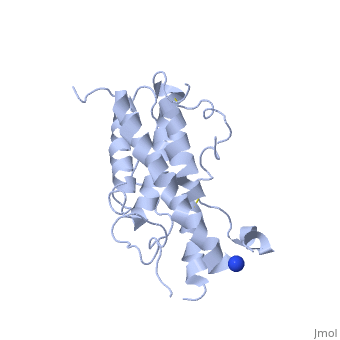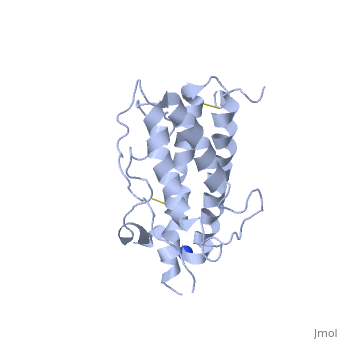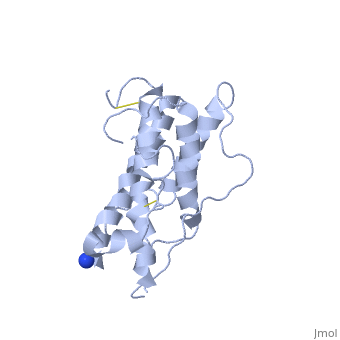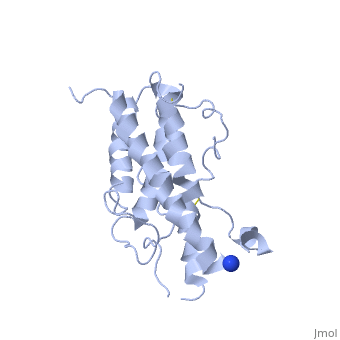
hGH is a single chain peptide, which was 45% helical with 8 α-helices (Chantalat et al., 1995)[4]. Protein Data Bank (PDB) provides predicted secondary structures of different proteins using the research from the paper and an algorithm program.
Tertiary Structure
The known crystal structure of hGH illustrate that the core of the protein is a four-helix bundle. Helices 1 & 4 at the NH2 and COOH ends are longer than helices 2 & 3 (Abraham 1992). A short loop connects helices 2 & 3 and two long crossover connections link helices 1 & 2 and helices 3 & 4. The helices run up-up-down-down (Abraham 1992). This is unusually because normally four helix bundles exhibit up-down-up-down orientation.
An additional three much smaller helices exist within the connecting loops: one at beginning of the connection between helices 1& 2, one at the end of the connection between helices 1 & 2, and one in the short loop connection between helices 2 & 3 (Abraham 1992).
Post-translational modifications
Phosphorylation
HGH is phosphorylated on Ser residues 132 and 176 (Giorgianni, Beranova-Giorgianni, and Desiderio, 2004)[5]. Other research has indicated possible phosphorylation of Tyr residues 35 and 42. However, these phosphorylations were only investigated in carcinoma cells with constitutively active epidermal growth factor-stimulated tyrosine kinase (Baldwin et al., 1983)[6]. The overall influence of these post-translational modifications on hGH activity has yet to be determined.
Glycosylation
Glycosylation helps distinguish between different variants and isoforms as it works as an ID cards for proteins. These carbohydrates are specific to each forms and are recognized by the associated hgH receptors. Though still being a relatively unknown mechanism in hgH, studies have shown that one isoform in particular, a 22kDa variant was identified and discovered due to the specific carbohydrates linked to its polypeptide chain (M.Kirstein 1992).
Secretion Mechanism
It was previously believed that after stimulating HGH secretion, membrane bound secretory vesicles containing the hormone dock at the cell plasma membrane. These vesicles were believed to become completely incorporated into the plasma membrane, and would later be retrieved via endocytosis, thus allowing for passive release of the HGH within the vesicles. However, this mechanism is not supported by experimental evidence, such as the appearance of empty and partially empty vesicles immediately after secretion. Bhanu Jena’s laboratory has recently elucidated the molecular mechanism of cellular secretion. Their studies suggest that there is actually a new cellular structure called a porosome that is involved with the mechanism. Porosomes are “basket-like” structures residing at the plasma membrane that have a 100-150nm diameter opening to the extracellular environment (Figure 1). It involves several specific proteins like SNAP receptors or N-ethylmaleimide-sensitive factor. First, the GH is brought to the porosome using ATP and kinesins along microtubules. Then, rather than docking directly at the plasma membrane post secretion stimulation, membrane bound secretory vesicles fuse at the base of porosomes, which subsequently expel the vesicular contents (Figure 2). During stimulation, the opening dilates about 20-35% to aid in the expulsion of HGH. The porosome returns to the resting size once the process is complete (Anderson et al., 2004)[7].
Several hypothalamic hormones that control growth hormone release exist. The most major of these secretion stimuli is the growth hormone release factor (GHRH). Another factor has been discover : ghrelin is an acylated peptide directly responsible for a GH secretion from somatotropes. In contrast, the hormone known as somatostatin (SRIF) is known to suppress the release of GH by the somatotrope. These two hormones are the most well known, though it should be noted that there are multiple other control factors which both stimulate and suppress the release of HGH into the bloodstream (Anderson et al., 2004).
Mechanism of Action
In order to facilitate this behavior as a hormone somatotropin binds to two receptors on the outside of a cell known as Human Growth Hormone Binding Proteins (hGHpb). Once the Human Growth hormone binds both receptors (first one, then the second), it causes a shift in the receptor protein, which in turn causes an internal signaling cascade. This cascade is how somatotropin is able to effect cell growth and function. In addition it can cause the release of other growth factors, like Insulin Growth Factor.
Examples :
For Growth :
HGH is especially important for the growth of cartilage and bone. It’s efficiency will increase even more during the adolescent years when it is more produced.
Insulin-like growth factor-1 binds to its receptor, IGF-1R, on the cellular surface and activates a tyrosine kinase-mediated intracellular signaling pathway that phosphorylates various proteins intracellularly leading to increased metabolism, anabolism, and cellular replication and division. Furthermore, it acts to inhibit apoptosis of the cell, thus prolonging the lifespan of existing cells. The net result is to encourage the growth of tissue and to create a hyperglycemic environment in the body.
Inhibitors
Somatostatin
The body’s primary mechanism for regulating hGH is to release somatostatin, also known as growth hormone inhibitory hormone (GHIH). Somatostatin is produced in the hypothalamus and released by the anterior pituitary gland, pancreas, and GI tract (Somatostatin, 2011). This hormone works together with growth hormone releasing hormone (GHRH) to properly regulate the secretion of hGH from the pituitary gland. Somatostatin levels are directly affected by levels of circulating hGH. Specifically, levels of somatostatin are high when hGH concentrations are high, and low when hGH is low.
There are two forms of somatostatin found in the body. One form, known as SS-14, is 14 amino acids long and found primarily in the nervous system and pancreas. The other form has an amino acid chain length of 28 and is called SS-28. This form is found predominantly in the GI tract. SS-28 is a much stronger inhibitor of hGH then SS-14 (Bowen, 2002). Most of the body’s receptors do not differentiate between the two forms of somatostatin. All receptors are G protein-coupled receptors and inhibit adenyl cyclase, which, in turn, affects a number of hormones and second messengers (Bowen, 2002).
Glucocorticoids
Glucocorticoids are a type of steroid hormones that regulate hGH levels in two different ways. Studies have shown that glucocorticoids are able to suppress the release of GHRH as well as reduce GHRH receptor responsiveness (Miller et al., 1997)[8]. Due to the suppression of GHRH, the levels of hGH in the body eventually subside. There is also data to suggest that glucocorticoids work with somatostatin release in the hypothalamus (Lima et al., 1993)[9].
Besides interactions with GHRH and somatostatin, glucocorticoids can affect adrenergic receptors (both alpha 2-adrenergic and beta-adrenergic receptors, which are stimulated by catecholamines in the sympathetic nervous system. When stimulated, the alpha receptors have shown to stimulate hGH release, whereas stimulation of beta receptors can inhibit hGH release (Blackard, 1968).
Hyperglycemia and Insulin
Periods of high sugar levels in the body are generally accompanied with higher insulin levels. In this state, insulin and related hormones like Insulin-like Growth Factor (IGF-1) have been shown to decrease binding affinity between hGH and its receptors (Shaonin et al., 1997). With lower levels of insulin and IGF-1, hGH secretion and levels can quickly and continuously rise. Although insulin and IGF-1 don't directly act on hGH receptors, they can affect the signaling cascade pathway that hGH uses (Yakar et al., 2004)[10]. JAK2 is one of the proteins found in this signaling pathway and it has been known to be affected during hGH/insulin feedback and regulation loops (Shaonin et al., 1997).
Associated Diseases and Treatments
Diseases associated with human growth hormone are related to deficiency or overproduction of the hormone.
Deficiency in somatostatin can occur in childhood or adulthood. Congenital deficiency is typically associated with an abnormal pituitary gland but can also be part of a larger syndrome or condition1. Acquired deficiency can develop from multiple sources including infection, brain tumors, injury, brain surgery or radiation to the head1.Pituitary tumors which have been treated with surgery or radiation are the typically cause of GH deficiency in adults.
Symptoms of deficiency in children include short stature, slow growth, late onset of puberty, increased fat around the waist, and delayed tooth development1. Pituitary dwarfism can result from untreated deficiency in children. In adults the symptoms include decreased strength and muscle mass, weight gain, anxiety and can increased total cholesterol and triglyceride levels.
The Turner syndrome is classified as a type of dwarfism affecting girls. It is a chromosomal disorder due to a partial or complete absence of an X chromosome.
The general treatment for dwarfism is HgH injections. The sooner this treatment is started, the more effective it will be. These injections are applied anywhere from several times a week to once a day.
Injection of GH has been implicated in the development of Creutzfeldt-Jakob Disease. Several cases of such disease were proven related to subjects being recipient of pituitary derived hGH. This association has only been found in GH isolated from cadavers. Originally isolation of the protein from cadavers was the method of development for replacement therapies. Now recombinant methods of production are the main method for synthesis. There seems to be no association in recombinant DNA-produced GH and Creutzfeldt-Jakob disease2.
Diseases can also result from levels of GH being too high. The effects of excess GH vary based on age. Gigantism is excess of GH during childhood and Acromegaly is excess of GH during adulthood (after bone growth has stopped). Symptoms of gigantism include delayed puberty, headache, increased sweating, and weakness3. Symptoms of acromegaly vary slightly and include carpal tunnel syndrome, weakness, joint pain, sleep apnea, and unintentional weight loss4.
Excess GH release most often occurs because of a pituitary gland tumor3. It can also be due to Carney complex, McCune-Albright syndrome, multiple endocrine neoplasia type 1, and neurofibromatosis3. Treatment includes medications to decrease hormone release and, in severe cases, removal of the pituitary gland.
Acromegaly is most commonly treated with surgery. Removing the tumor in the pituitary gland can stop the excess release of growth hormone. Incomplete success of the surgery is often attributed to the size of the tumor: large tumors cannot always be completely removed, resulting in continued high levels of hormone release (Freda, 2002)[11].
Surgery is often supplemented, or in some cases replaced, by radiation therapy. Radiation of the pituitary can help reduce levels of hormone release by killing tumor cells, but reduction of release as a result of radiation is a slow process and is not as successful as surgery. For this reason, it is more commonly used in tandem with surgery, rather than alone (Freda, 2002; A.D.A.M. Acromegaly).
Certain medications can also be used in some combination with surgery and radiation. There are two classifications of acromegaly medication treatments. The first reduces HgH release from the pituitary. Somatostatin agonists reduce HgH release in approximately 50 percent of patients. They act by binding to somatostatin receptors, which regulate hormone control. The seemingly low level of success is due to certain tumors being resistant to somatostatin agonists. There are currently two somatostatin agonists on the market. Octreotide is a long acting release medication that needs to be injected about once a month. Lanreotide is a slow release medication that needs to be administered by injection about every other week. The second class of medication is an analog of HgH that competitively binds to HgH receptors, without activating them. Pegvisomant is an HgH analog that blocks the binding of HgH to HgH inhibitors. It is injected daily (Freda, 2002; A.D.A.M. Acromegaly).