We apologize for Proteopedia being slow to respond. For the past two years, a new implementation of Proteopedia has been being built. Soon, it will replace this 18-year old system. All existing content will be moved to the new system at a date that will be announced here.
Investigating the Mechanisms of Active Site Mutations to the 1T9G WT MCAD Protein to Better Understand Medium Chain Acyl-CoA Dehydrogenase Deficiency (MCADD)
From Proteopedia
(Difference between revisions)
| Line 5: | Line 5: | ||
== Abstract == | == Abstract == | ||
| - | Medium-chain Acyl-CoA Dehydrogenase Deficiency (MCADD) is a human disorder that hinders β-oxidation, affecting approximately 1 in 17,000 people in the United States. Once mutated, the Acyl-CoA Dehydrogenase Medium-Chain (ACADM) gene, which is solely responsible for MCADD, cannot produce enough MCAD enzymes to metabolize medium-chain fatty acids. As a result, fats are not catabolized, causing symptoms of lethargy and hypoglycemia, as well as damage to the brain and liver due to a buildup of unused fatty tissue. The purpose of this project was to investigate the possible and known effects of different amino acid mutations on the human MCAD protein and produce a 3D-printed model to explain the molecular story of MCADD. This model builds on previous bioinformatics and in vivo experiments aimed at revealing the underlying enzymatic mechanisms of MCADD. Using PyMOL, the human wild-type MCAD (PDB ID: 1T9G) had its electron-transferring flavoprotein (ETF) complex removed and a single chain from its homotetramer portion isolated for clarity. PyRx was used to dock the substrate, Octanoyl-CoA (PDB ID: CO8) into the slightly mutated enzyme, referencing PDB ID 1EGC. Known mutations from the PDB files and related literature were then compared and analyzed on the modified 1T9G to determine known and possible effects the mutations had, such as helix-helix stability and ligand hydrogen bonding. LigPlot+ was then used to analyze ligand-active site interactions. Jmol was used to cosmetically enhance the modified 1T9G to produce a 3D model for printing. In the model, the mutations were ranked according to known KM range values (0.4, 0.6), (0.7, 0.9), and 0.9+, which were highlighted in the colors “lightskyblue”, “royalblue”, and “midnightblue”, respectively; unknown KM values were colored “chartreuse”. All mutations had their side chains shown for further clarity. E376, the catalytic base, was colored “magenta”, the backbone was colored “dimgray”, and the support struts of the model were colored “lightseagreen”. The use of the 3D model was beneficial, enabling model viewers to locate, determine, and hypothesize the mutations and their effects on MCAD, in addition to providing a visual and physical learning aid for researchers, professors, students, and other biomedical professionals. Furthermore, the clarity produced by a physical model ultimately enables further research for MCADD and may assist in the development of a cure for those who unfortunately suffer from this rare condition. | + | Medium-chain Acyl-CoA Dehydrogenase Deficiency (MCADD) is a human disorder that hinders β-oxidation, affecting approximately 1 in 17,000 people in the United States <ref>https://medlineplus.gov/genetics/condition/medium-chain-acyl-coa-dehydrogenase-deficiency/</ref>. Once mutated, the Acyl-CoA Dehydrogenase Medium-Chain (ACADM) gene, which is solely responsible for MCADD, cannot produce enough MCAD enzymes to metabolize medium-chain fatty acids. As a result, fats are not catabolized, causing symptoms of lethargy and hypoglycemia, as well as damage to the brain and liver due to a buildup of unused fatty tissue. The purpose of this project was to investigate the possible and known effects of different amino acid mutations on the human MCAD protein and produce a 3D-printed model to explain the molecular story of MCADD. This model builds on previous bioinformatics and in vivo experiments aimed at revealing the underlying enzymatic mechanisms of MCADD. Using PyMOL, the human wild-type MCAD (PDB ID: 1T9G) had its electron-transferring flavoprotein (ETF) complex removed and a single chain from its homotetramer portion isolated for clarity. PyRx was used to dock the substrate, Octanoyl-CoA (PDB ID: CO8) into the slightly mutated enzyme, referencing PDB ID 1EGC. Known mutations from the PDB files and related literature were then compared and analyzed on the modified 1T9G to determine known and possible effects the mutations had, such as helix-helix stability and ligand hydrogen bonding. LigPlot+ was then used to analyze ligand-active site interactions. Jmol was used to cosmetically enhance the modified 1T9G to produce a 3D model for printing. In the model, the mutations were ranked according to known KM range values (0.4, 0.6), (0.7, 0.9), and 0.9+, which were highlighted in the colors “lightskyblue”, “royalblue”, and “midnightblue”, respectively; unknown KM values were colored “chartreuse”. All mutations had their side chains shown for further clarity. E376, the catalytic base, was colored “magenta”, the backbone was colored “dimgray”, and the support struts of the model were colored “lightseagreen”. The use of the 3D model was beneficial, enabling model viewers to locate, determine, and hypothesize the mutations and their effects on MCAD, in addition to providing a visual and physical learning aid for researchers, professors, students, and other biomedical professionals. Furthermore, the clarity produced by a physical model ultimately enables further research for MCADD and may assist in the development of a cure for those who unfortunately suffer from this rare condition. |
== Introduction == | == Introduction == | ||
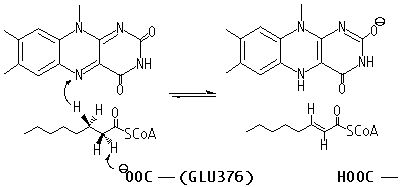
| - | An important enzyme in β-oxidation is Acyl-CoA Dehydrogenase, which abstracts a hydrogen atom from its fatty acyl-CoA substrate and inserts it on FAD, an electron carrier. With FAD also removing a fatty acyl-CoA hydrogen, FAD is reduced to FADH2, which is utilized in the electron transport chain to ultimately produce ATP, forming a double bond on the acyl-CoA chain. The biochemical mechanism is shown in the image below<ref>https:// | + | An important enzyme in β-oxidation is Acyl-CoA Dehydrogenase, which abstracts a hydrogen atom from its fatty acyl-CoA substrate and inserts it on FAD, an electron carrier. With FAD also removing a fatty acyl-CoA hydrogen, FAD is reduced to FADH2, which is utilized in the electron transport chain to ultimately produce ATP, forming a double bond on the acyl-CoA chain. The biochemical mechanism is shown in the image below<ref>Bach, R. D., Thorpe, C., & Dmitrenko, O. (n.d.). Synergy Between H-Bonding Interactions and Its Role in the Enzyme-Catalyzed a-Proton Abstraction. DFT Studies On the Acyl-CoA Dehydrogenase Model Systems. University of Delaware. https://www1.udel.edu/chem/bach/pages/CCE8corr.html</ref>. In Medium Acyl-CoA Dehydrogenase Deficiency (MCADD), mutations in the ACADM (Acyl-CoA Dehydrogenase Medium-Chain) gene, the only gene that causes MCADD <ref>Drendel, H. M., Pike, J. E., Schumacher, K., Ouyang, K., Wang, J., Stuy, M., Dlouhy, S., & Bai, S. (2015). Intermediate MCAD Deficiency Associated with a Novel Mutation of the ACADM Gene: c.1052C>T. Case reports in genetics, 2015, 532090. https://doi.org/10.1155/2015/532090</ref>, render less functional MCADs. Since MCADD is the most common defect in the pathway of β-oxidation, and MCAD (medium-chain acyl-CoA dehydrogenase) is needed to metabolize medium-chain fatty acids, a deficiency of this protein has effects ranging from hypoglycemia and lethargy, and damage to the brain and liver due to a buildup of fatty tissue <ref>Drendel, H. M., Pike, J. E., Schumacher, K., Ouyang, K., Wang, J., Stuy, M., Dlouhy, S., & Bai, S. (2015). Intermediate MCAD Deficiency Associated with a Novel Mutation of the ACADM Gene: c.1052C>T. Case reports in genetics, 2015, 532090. https://doi.org/10.1155/2015/532090</ref>. Understanding of the mutations that caused the disease was sought; amino acid mutations that overlapped across the studies researched and were able to be visualized in the Human WT MCAD (PDB ID: 1T9G) were recorded and analyzed for their effects on the protein (i.e., helix-helix interactions, H-bonding to ligand) and how it could contribute to MCAD; these mutations are listed in the colored table to the below. |
| - | https:// | + | https://www1.udel.edu/chem/bach/pages/Image1.gif |
== Materials & Methods == | == Materials & Methods == | ||
Revision as of 20:51, 21 May 2023
Investigating The Mechanisms of Active Site Mutations to the 1T9G WT MCAD Protein to Better Understand Medium Chain Acyl-CoA Dehydrogenase Deficiency (MCADD) [1]
| |||||||||||
References
- ↑ Saleh, Omar E.; Khatiwala, Rhea; and Ignatius, Jeremy, "Investigating The Mechanisms of Active Site Mutations to the 1T9G WT MCAD Protein to Better Understand Medium Chain Acyl-CoA Dehydrogenase Deficiency (MCADD)" (2022). Protein Modeling Reports. 7. https://nsuworks.nova.edu/protein_modeling_reports/7
- ↑ https://medlineplus.gov/genetics/condition/medium-chain-acyl-coa-dehydrogenase-deficiency/
- ↑ Bach, R. D., Thorpe, C., & Dmitrenko, O. (n.d.). Synergy Between H-Bonding Interactions and Its Role in the Enzyme-Catalyzed a-Proton Abstraction. DFT Studies On the Acyl-CoA Dehydrogenase Model Systems. University of Delaware. https://www1.udel.edu/chem/bach/pages/CCE8corr.html
- ↑ Drendel, H. M., Pike, J. E., Schumacher, K., Ouyang, K., Wang, J., Stuy, M., Dlouhy, S., & Bai, S. (2015). Intermediate MCAD Deficiency Associated with a Novel Mutation of the ACADM Gene: c.1052C>T. Case reports in genetics, 2015, 532090. https://doi.org/10.1155/2015/532090
- ↑ Drendel, H. M., Pike, J. E., Schumacher, K., Ouyang, K., Wang, J., Stuy, M., Dlouhy, S., & Bai, S. (2015). Intermediate MCAD Deficiency Associated with a Novel Mutation of the ACADM Gene: c.1052C>T. Case reports in genetics, 2015, 532090. https://doi.org/10.1155/2015/532090
- ↑ Toogood, H. S., van Thiel, A., Basran, J., Sutcliffe, M. J., Scrutton, N. S., & Leys, D. (2004). Extensive domain motion and electron transfer in the human electron transferring flavoprotein·medium chain acyl-COA dehydrogenase complex. Journal of Biological Chemistry, 279(31), 32904–32912. https://doi.org/10.1074/jbc.m404884200
