Ashley Callaghan/Sandbox1
Introduction
Leaf branch compost cutinase is a versatile enzyme that can break down both natural plant polymers and synthetic plastics.[1] It was discovered in a compost heap, and it originally evolved to degrade cutin, the protective biopolymer in plant surfaces. LCC has also shown high efficiency in hydrolyzing polyethylene terephthalate (PET), which is a widely used plastic that contributes to global pollution. Unlike many other PET-degrading enzymes, LCC is thermostable and has a high catalytic efficiency, which means it can function at temperatures that are optimal for industrial recycling processes. By breaking PET into its monomers, LCC promotes closed-loop recycling of plastic waste and reduces environmental accumulation.
[2]
[3]
Function
LCC catalyzes the hydrolysis of the ester bonds in PET polymers and breaks them down into their constituent monomers: terephthalic acid and ethylene glycol. The enzyme operates through a catalytic triad that consists of S165, D210, and H242. Using these residues, LCC can perform nucleophilic attacks on the carbonyl carbon atoms of the ester bonds in PET. During catalysis, the substrate binds in an elongated, predominantly hydrophobic groove present in the enzyme's structure.
LCC functions optimally at elevated temperatures (around 65-72°C), which approaches the glass transition temperature of PET. This temperature range maximizes PET chain mobility and makes the polymer more accessible to enzymatic action. The enzyme is remarkably thermostable compared to other PET hydrolases, with a melting temperature of 84.7°C. This property allows it to remain functional under these high-temperature conditions. Also unlike other PET hydrolases such as Is-PETase, BTA1, BTA2, and FsC, LCC has substantially higher catalytic efficiency. Specifically, LCC has an initial PET-specific depolymerization rate of 93.2 mg TAeq. h⁻¹ mg⁻¹ enzyme at 65°C with amorphous PET. This means that it us at least 33 times more efficient than other tested enzymes.[1]
LCC's function is limited by PET crystallinity, as the enzyme can more effectively hydrolyze amorphous regions of the polymer. As PET crystallinity increases during the depolymerization reaction (due to exposure to elevated temperatures), the enzyme's efficiency decreases. This limits complete depolymerization unless optimal conditions and enzyme variants are used.
Relevance
With global plastic production reaching approximately 299 million tons annually, the need for effective waste management solutions is urgent. Enzymatic degradation is an alternative to conventional recycling methods that are often inefficient and taxing on the environment. One of the primary challenges in plastic waste management is the volume of mismanaged plastic entering marine environments. In 2010 alone, an estimated 31.9 million metric tons of plastic waste were classified as mismanaged, with a substantial portion ending up in the oceans. This causes harm to marine ecosystems, physical injury to wildlife, and disruption of food chains.[4]
Integrating LCC into existing waste management systems could substantially reduce the PET waste that enters the environment. Research suggests that a 77% reduction in mismanaged plastic waste could lower the annual input of plastic into the ocean to between 2.4 and 6.4 million metric tons by 2025.
LCC hydrolyzes PET into its constituent monomers, which also supports the principles of a circular economy, where materials are reused rather than discarded. Enzymatic degradation allows for the production of biologically recycled PET with properties that are comparable to virgin materials.[5]
Structural Overview
Catalytic Triad
LCC catalyzes the breakdown of PET using a classic serine hydrolase mechanism involving a of Ser165, His242, and Asp210. The reaction begins when His242 deprotonates Ser165, which activates it as a nucleophile. Ser165 then attacks the carbonyl carbon of an ester bond in the PET polymer. This forms a tetrahedral intermediate. This intermediate is stabilized by an oxyanion hole. The oxyanion hole is formed by the backbone amides of Met166 and Tyr95. The intermediate collapses; one product is released and an acyl-enzyme intermediate is formed. A water molecule, activated by His242, then attacks the acyl-enzyme, releasing the second product and resetting the enzyme’s active site.
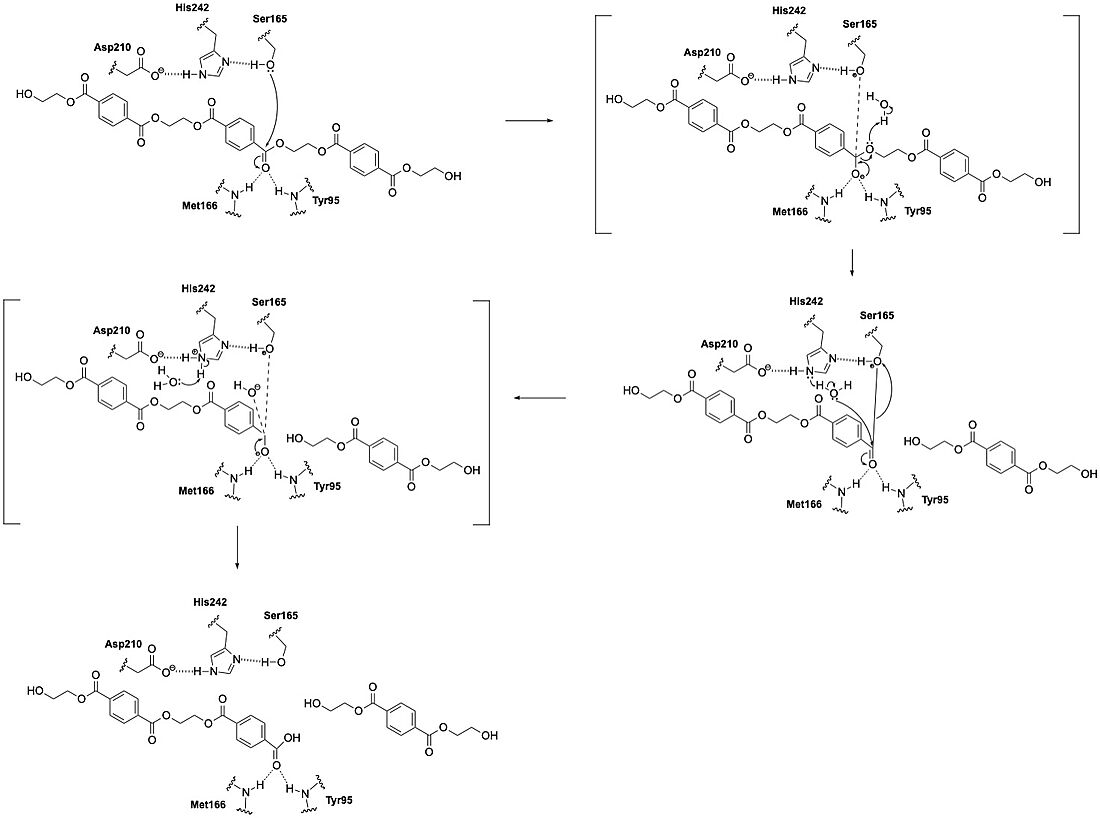
Ligand Binding Pocket
The of LCC is a long, mainly hydrophobic groove that accommodates PET chains. This groove includes three subsites—designated −2, −1, and +1—that interact with specific PET units near the scissile ester bond. Hydrophobic residues such as F125, V212, M166, and F243 line the groove and facilitate binding by interacting with the aromatic rings of the PET molecule. These interactions help align the substrate in the correct position for catalysis.
Mutation Sites of Interest
F243
F243W
The original side chain is phenylalanine side chain, is located 3.6 Angstroms from the ligand. There are two main mutations isoleucine and tryptophan. Isoleucine is a smaller molecule and makes a tighter bond in the ligand, with the distance being 3.0 Angstrom. This allows for more efficient interactions with the ligand, likely improving substrate binding and catalytic efficiency. The other is tryptophan, which is a bulkier side chain, which does have a slightly shorter difference of 3.2 Angstroms, while being a larger side chain. The effect that is has is more based on that tryptophan is a nitrogen-containing aromatic ring, which offers new interactions with the substrate, which could also affect enzyme's binding and catalytic properties.
These two mutations both lead to an increase in catalytic activity from the mutation, F243I had a 27.5% increase and F243W had an 17.5% increase, when compared to the wild-type variant.
T96
T96M
The original side chain is threonine and is mutated with a methionine. This is really a mutation that increases thermostability. The wild-type variant has a melting point of 84.7 degree Celsius. The mutation of methionine, is a larger, more hydrophobic side chain, and would contribute to stabilization of the protein's core by increasing hydrophobic interactions. The stronger internal interactions help the enzyme to maintain its folded structure more efficiently at elevated temperatures. Which is shown by the M96 mutation having a melting point of 87.4 degree Celsius.
Y127
The original side chain is tyrosine, and is has a mutation to glycine. Like the T96M mutation, it also increases thermostability of the protein, with this mutation Y127G having a melting point of 87.0 degree Celsius. The mutation replaces tyrosine, a bulky, rigid aromatic side chain to glycine, which is the smallest amino acid. This mutation leads to allowing greater flexibility for the protein. By reducing steric hindrance and releasing some strain in the protein structure, allowing for it to be more flexible and more stable at higher temperatures.
N246
N246D
N246M
The asparagine side chain is mutated to aspartic acid and methionine to increase thermostability. The wild-type protein has a melting point of 84.7°C. The N246D mutation (asparagine to aspartic acid) replaces a polar neutral side chain with a negatively charged one, which potentially increases electrostatic interactions or salt bridges that stabilize the protein. This results in a melting point of 87.9°C. The N246M mutation (asparagine to methionine) introduces a bulkier, hydrophobic side chain. This increases the internal packing of the protein core. This mutant has a melting point of 88.0°C. These mutations are designed to improve the protein's performance in plastic degradation by increasing its thermal stability.
S283 & D238
These are two mutations that are linked one being a serine and aspartic acid. These mutations were meant to replace calcium ions which are very common in these experiments, in an attempt to replace them with a disulfide bond. As their distances between the alpha and beta atoms suggesting that it could be engineered. With the mutation, of both to a cystine, allowing for disulfide bonds, found improved thermostability. Wild-type having a melting point of 84.7 degree Celsius. While the mutation was highly successful in increased thermostability, with the mutation having a melting point of 94.5 degree Celsius, which is a 9.8 degree Celsius increase, which is higher than the rest of the mutations. However, this was also shown to have a result of a decrease in enzymatic activity of 28% compared to wild type.
This is likely why many mutations are all used at once, in order to increase thermostability and maintain or increase catalytic activity. Since they are all for different sites that are mutated, they can be done in combination, depending on what you are looking for. It will also be very important for expanding the look into other substrates and enzymes.


