Introduction
The Diels-Alderase catalyzes the Diels-Alder reaction [Fig. 1A] between 4-carboxybenzyl-trans-1,3-butadiene-1-carbamate and N,N-dimethylacrylamide [Fig. 1B] for use in synthetic organic chemistry. Specifically, the enzyme surpasses uncatalyzed reactions by generating a product that is entirely stereoselective for the 3R,4S endo form [Fig.1C]. Though other attempts at controlling Diels-Alder steroisomerism had been attempted,[1] this project was the first to use an enzyme model. The Diels-Alderase was built using de novo enzyme design, using computational modeling and refinement through collaborative problem-solving from online users. The first generation Diels-Alderase was made using the Rosetta computational design program, where a potential active site was built and tested against a library of scaffold proteins. Later, as the active site was perfected, future generations of the Diels-Alderase were made using an online protein folding game called Foldit, where players competed to improve binding efficiency by completing various challenges.[2]
The Diels-Alderase was designed to connect a diene and dienophile to complete the Diels-Alder reaction. It accomplishes this by decreasing the energy gap between the dienophile’s lowest unoccupied molecular orbital (LUMO) and the diene’s highest occupied molecular orbital (HOMO) in the transition state.[3] The current most active form of the Diels-Alderase is modelled under the PDB code 4o5t.
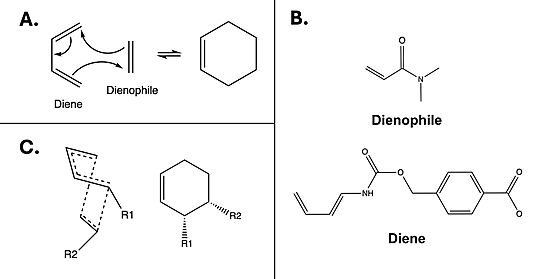
Figure 1. A) Example mechanism of a simple Diels-Alder reaction. B) Diels-Alderase substrates. Diene is 4-carboxybenzyl trans-1,3-butadiene-1-carbamate; dienophile is N,N- dimethylacrylamide. C) Illustration of 3R, 4S endo stereoisomerism, which the Diels-Alderase is selective for.
The binding pocket of 4o5t is selective for two substrates, 4-carboxybenzyl trans-1,3-butadiene-1-carbamate (diene) and N,N- dimethylacrylamide (dienophile). These substrates are shown as a single, combined ligand, 4-{[2-(phosphonooxy)ethyl]carbamoyl}benzyl [(1R,6S)-6-(dimethylcarbamoyl)cyclohex-2-en-1-yl]carbamate, in the protein model. The binding site contains a hydrogen bond donor (Tyr134) which lowers the LUMO energy and stabilizes the negative charge on the dienophile.[3] It also contains a hydrogen bond acceptor (Glu208) that increases the HOMO energy and stabilizes the positive charge on the diene.[3] Both of these H-bonding interactions work to stabilize the transition state while also orienting the substrates in optimal conformations for reacting.
Overall, the Diels-Alderase stimulates improvement in synthetic laboratories and demonstrates early success in the now-prominent world of computational enzyme design.
General Structure
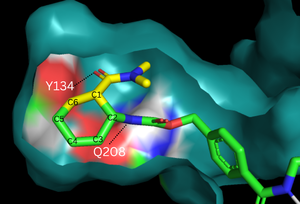
Figure 2. Binding pocket and substrate. Shown is the binding pocket of the enzyme shown as surface, highlighting the electrostatics of the two catalytic residues, Tyr134 and Glu208. The ligand is color coded based on original structure: the dienophile is in yellow and the diene is in green. The reaction proceeds via attack of the C6 on the C5, shifting electron density to C2, which attacks C1.
Scaffold
The original enzyme scaffold was found using Rosetta parameters that searched for enzymes that contained catalytic Tyr and Glu residues and could coordinate two substrates highly specifically. Using the software, a catalog of 207 scaffolds was screened for potential active site orientations that could accommodate the substrates. 84 of the 207 were selected for testing, 50 were found to be soluble, and only 2, DA_20_00 and DA_42_00, had any enzymatic activity.[3] Preliminary results favored and thus this beta-propeller scaffold was chosen as the base for further experiments.[3][4] The DA_20_00 protein scaffold is a 6-bladed beta-propeller of Loligo vulgalis, or the European Squid. The protein is relatively simple, with only one chain, one unit, 324 residues, and no extra ligands, metal ions, or small molecules bound.
Active Site
In the designed active site, stabilize the transition state of the Diels-Alder reaction. The Tyr134 acts as a to the oxygen on the dienophile [Fig. 2]. Q208 acts as a to the nitrogen on the diene [Fig. 2]. These interactions help reduce the energetic gap between orbitals, allowing the reaction to proceed. The active site geometry also plays a large role in the binding of the substrates and how they react on a stereochemical level. By making small changes in the active site, changes can be made to the selectivity.
Helix Cap
In the evolution process, a 16-residue alpha-helix to the top of the binding site. The original hypothesis was that including a steric group near to the top of the active site would increase the binding affinity of the enzyme and improve the reaction kinetics. It was experimentally shown that he hydrophobic helix “functions as a lid to constrain the substrates in a productive orientation for reaction,” decreasing the Km of the enzyme and increasing the catalytic efficiency, as seen in the measured kinetics of the enzyme.[2]
Mechanism
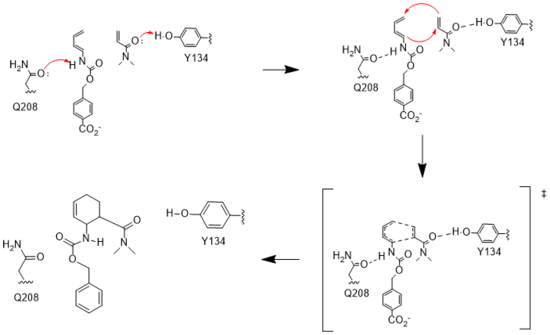
Figure 3. Active site mechanism
The key to the Diels-Alderase's success as a catalyst lies in its ability to lower the energy gap between reactants. To accomplish this, the two active site residues, Tyr134 and Glu208, use hydrogen bonding to assist the reaction in a variety of ways.
First, it allows specific binding of the ligand in the active site, selecting for molecules with certain stereochemistry at and around the catalytic residues, specifically the carbamate and carbonyl of the diene and dienophile, respectively. This promotes the reaction by stabilizing the molecules in close proximity to one another, also promoting the reaction's characteristic stereoselectivity.[3] Second, the bonds affect the energetics of the molecules. By donating a hydrogen to the carbonyl of dienophile, Tyr134 helps to decrease the electron density around the molecule, lowering the energy of the lowest unoccupied molecular orbital (LUMO).[3] Conversely, by abstracting the hydrogen from the carbamate of the diene, Glu208 increases the electron density and thus the energy of the highest occupied molecular orbital (HOMO). By closing the gap between these orbitals, the enzyme lowers the activation energy required for the orbitals to react. Finally, these interactions help to stabilize the accumulated charges in the transition state. By decreasing electron density in the dienophile, Tyr134 helps to stabilize the accumulated negative charge in the transition state. The Glu208, then, helps stabilize the accumulated positive charge by increasing the electron density of the diene. Calculations predict that this helps to stabilize the transition state by nearly 5 kcal/mol.[3] All together, these interactions make it much easier for the reaction to proceed in a very stereoselective and favorable manner.
Development and Evolution
DA_20_00
During initial computer modelling, over one million potential Diels-Alderase active sites were matched to potential protein scaffolds.[3] Only 2 proteins proved to be sufficiently active after LC-MS screening. DA_20_00, which used a beta-propeller scaffold, had the most success in further mutations and therefore became the Diels-Alderase of choice.[3] However, this initial enzyme's active site had very little catalytic activity, seen in its low catalytic efficiency after kinetic screening [Fig. 4].[3][5]
DA_20_10
DA_20_10 provided key mutations in and around the active site that increased the hydrophobicity, provided structural stability, and increased interactions between the ligand and surrounding residues.
Q162R
- Glu162, a , resides near the top of the binding site, and is about than 3Å from the ligand in most models on the enzyme. It can act as a hydrogen bond donor to the terminal phosphate on the ligand when in proximity. To increase this interaction, Glu162 was mutated to an , which decreased the length of the potential hydrogen bond to within 2.5Å in most models, increasing the strength of the interaction.[3]
S284A
- Ser284 resides deep within the binding pocket of the enzyme. Choosing a to mutation increases the hydrophobicity of the binding pocket and reduce reactivity, without also changing any steric characteristics in the region unintentionally near the catalytic residues.[3]
A285N
- Introducing an to increases steric hindrance with the catalytic tyrosine, reducing the number of rotamers the residue has to increase the reactivity of the enzyme by lowering the distance between Tyr134 and the ligand.[3]
CE6
The DA_20_10 model of the Diels Alderase was further enhanced by players of the online game Foldit.[2] Building on preliminary early data, players were asked to optimize various helical structures that would surround and support the ligand. After over 100,000 designs were tested, the top-scoring CE6 model was finalized, containing the that favorably constrains ligand orientation. This "cap" consists of two helices--helix one spans from residues 36-44, and helix two spans from residues 48-56.[2]
CE20
The CE20 generation contains three highly conserved mutations found in many of the most catalytically efficient Diels-Alderase models being screened: Tyr43, Pro48, and Arg56 were mutated to .[5] Generally, these mutations each contributed to further tightening the binding pocket around the ligand and creating a more hydrophobic environment for enhanced binding.[5]
Kinetics
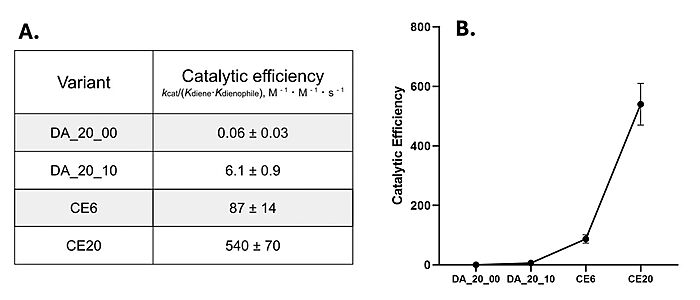
Figure 4. A) Catalytic efficiencies of key Diels-Alderase generations. Kinetic data was measured at 25°C, in PBS, at pH 7.4. B) Improvement of catalytic efficiency across generations.
[5]Classic Michaelis-Menten kineticswere determined for each generation of the enzyme. As the Diels-Alderase relies on a catalyzed interaction between both the diene and dienophile, a Michaelis binding constant (Km value) was determined for each substrate separately before catalytic efficiency was calculated. The CE20 model of the enzyme is over 300-fold more efficient than the first enzyme model due to increasing active site specificity [Fig. 4].[5] The authors did not publish comparisons to free reflux originally.
Applications
The CE20 model is the most efficient Diels-Alderase yet, surpassing many other biological (antibody[1]) and artificial (ribozyme, metalloenzyme) attempts at catalyzing the Diels-Alder reaction.[5] Even then, the CE20 model has a catalytic efficiency value at least 4 orders of magnitude lower than the preferred values seen in any moderately-efficient natural enzymes catalyzing various reactions. This demonstrates the innate slowness of the Diels-Alder reaction.[5]
Though the rate of product formation using this enzyme is not significantly different from that found when reactants reflux free in solution (about 10 substrate molecules/hour),[5] the Diels-Alderase shows modest (15% [6]) improvement in product stereoselectivity. When refluxed in a room temperature aqueous solution containing the necessary substrates, the enzyme catalyzed an over 90% conversion rate, producing only the 3R,4S endo cyclohexane product isomer. By comparison, refluxing the substrates free in an aqueous PBS buffer at 110 °C for a similar duration of time yields an 85:15 mixture of endo and exo products.[6] It is primarily for these stereoselective benefits that this enzyme is valuable for synthetic purposes. [5]
Future improvement of the Diels-Alderase will likely revolve around the selective production of varying stereoisomers as well as improvement of catalytic efficiency and further constriction of the active site. Furthermore, the methodology and success behind designing the Diels-Alderase demonstrated the utility of early computational enzyme design, which is now an enormous field and the subject of the 2024 Nobel Prize.
References
- ↑ 1.0 1.1 Gouverneur VE, Houk KN, de Pascual-Teresa B, Beno B, Janda KD, Lerner RA. Control of the exo and endo pathways of the Diels-Alder reaction by antibody catalysis. Science. 1993 Oct 8;262(5131):204-8. PMID:8211138 doi:10.1126/science.8211138
- ↑ 2.0 2.1 2.2 2.3 Eiben CB, Siegel JB, Bale JB, Cooper S, Khatib F, Shen BW, Players F, Stoddard BL, Popovic Z, Baker D. Increased Diels-Alderase activity through backbone remodeling guided by Foldit players. Nat Biotechnol. 2012 Jan 22;30(2):190-2. doi: 10.1038/nbt.2109. PMID:22267011 doi:http://dx.doi.org/10.1038/nbt.2109
- ↑ 3.00 3.01 3.02 3.03 3.04 3.05 3.06 3.07 3.08 3.09 3.10 3.11 3.12 3.13 Siegel JB, Zanghellini A, Lovick HM, Kiss G, Lambert AR, St Clair JL, Gallaher JL, Hilvert D, Gelb MH, Stoddard BL, Houk KN, Michael FE, Baker D. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction. Science. 2010 Jul 16;329(5989):309-13. PMID:20647463 doi:329/5989/309
- ↑ Scharff EI, Koepke J, Fritzsch G, Lucke C, Ruterjans H. Crystal structure of diisopropylfluorophosphatase from Loligo vulgaris. Structure. 2001 Jun;9(6):493-502. PMID:11435114
- ↑ 5.0 5.1 5.2 5.3 5.4 5.5 5.6 5.7 5.8 Preiswerk N, Beck T, Schulz JD, Milovnik P, Mayer C, Siegel JB, Baker D, Hilvert D. Impact of scaffold rigidity on the design and evolution of an artificial Diels-Alderase. Proc Natl Acad Sci U S A. 2014 May 20. pii: 201401073. PMID:24847076 doi:http://dx.doi.org/10.1073/pnas.1401073111
- ↑ 6.0 6.1 Cannizzaro CE, Ashley JA, Janda KD, Houk KN. Experimental determination of the absolute enantioselectivity of an antibody-catalyzed Diels-Alder reaction and theoretical explorations of the origins of stereoselectivity. J Am Chem Soc. 2003 Mar 5;125(9):2489-506. PMID:12603137 doi:10.1021/ja020879d
Student Contributors
Taylor Donahue, Kate Thuma, Micah Zile




