This old version of Proteopedia is provided for student assignments while the new version is undergoing repairs. Content and edits done in this old version of Proteopedia after March 1, 2026 will eventually be lost when it is retired in about June of 2026.
Apply for new accounts at the new Proteopedia. Your logins will work in both the old and new versions.
Cyclooxygenase
From Proteopedia
| Line 1: | Line 1: | ||
| + | COX-1 and COX-2, also called PGHS-1 and PGHS-2, regulate a key step in prostaglandins and thromboxanes synthesis and are the targets of nonsteroidal antiinflammatory drugs (NSAIDs) (Smith & Langenbach, 2001; Ghosh et al., 2010; Chandrasekharan et al., 2002). Prostaglandins are implicated in various pathophysiological processes such as inflammatory reaction, gastrointestinal cytoprotection, hemostasis and thrombosis, as well as renal hemodynamics (Smith & Langenbach, 2001; Ghosh et al., 2010; Smith et al., 2000). Whereas COX-1 presents a widespread constitutive expression, COX-2 is undetectable in most normal tissues (except for the central nervous system, kidneys, and seminal vesicles), but is induced by various inflammatory and mitogenic stimuli (Smith et al., 2000; Ghosh et al., 2010; Rang & Dale, 2008). More recently, a third isoform named COX-3 was identified as a COX-1 splicing variant. This new isoform may play a role in fever and pain processes (Ghosh et al., 2010; Chandrasekharan et al., 2002). | ||
| + | Additionally, a high level of COX-2 expression is found usually in cancer cells (Ghosh et al., 2010). For example, COX-2 overexpression is related to poor-prognosis breast cancer (Boland et al., 2004; Barnes et al., 2007) and endometrial adenocarcinomas (Sales et al., 2008). | ||
| + | |||
==About this structure (1,2)== | ==About this structure (1,2)== | ||
PGHSs are bifunctional homodimers. Both COX-1 and COX-2 are membrane-bound enzymes and are present on the lumenal surfaces of the endoplasmic reticulum and of the inner and outer membranes of the nuclear envelope. However, recently, it has been demonstrated in cultured endothelial cells and fibroblasts that a fraction of COX-2 protein is localized to plasma membrane in caveolae-like structures (3). The primary structure of nascent COX-2 is of 604 amino acids and then it is processed into a mature form by removal of signal peptides giving a protein of 587 amino acids. PGHS-2 is variably glycosylated at two to four sites, leading to the formation of doublets or sometimes triplets on SDS-PAGE. Murine PGHS-2 peptide is presumed to be <scene name='SandboxUAM/Mynewscene/1'>N-glycosilated</scene> three times at Asn56, Asn130, and Asn396(NUEVA). | PGHSs are bifunctional homodimers. Both COX-1 and COX-2 are membrane-bound enzymes and are present on the lumenal surfaces of the endoplasmic reticulum and of the inner and outer membranes of the nuclear envelope. However, recently, it has been demonstrated in cultured endothelial cells and fibroblasts that a fraction of COX-2 protein is localized to plasma membrane in caveolae-like structures (3). The primary structure of nascent COX-2 is of 604 amino acids and then it is processed into a mature form by removal of signal peptides giving a protein of 587 amino acids. PGHS-2 is variably glycosylated at two to four sites, leading to the formation of doublets or sometimes triplets on SDS-PAGE. Murine PGHS-2 peptide is presumed to be <scene name='SandboxUAM/Mynewscene/1'>N-glycosilated</scene> three times at Asn56, Asn130, and Asn396(NUEVA). | ||
| Line 56: | Line 59: | ||
==NSAIDs== | ==NSAIDs== | ||
| + | |||
| + | Non-steroid anti-inflammatory drugs are a chemically heterogeneous group of compounds whose major function is the inhibition of cyclooxygenases (Table 1). Apart from their anti-inflammatory effect, they also present analgesic and antipyretic properties (Rang & Dale, 2008). | ||
| + | Classical NSAIDs, as salicylate or phenoprofen, are mostly inhibitors of both isoenzymes, although each isoform is inhibited in a different level. Chronic users of NSAIDs develop gastric ulcers or gastrointestinal complications, explained by the inhibition of COX-1. For this reason, selective inhibitors of COX-2, as celecoxib, valdecoxib and etoricoxib, have been developed (Ghosh et al., 2010, Rang & Dale, 2008). They don’t cause gastric pathology, but it has been proved to be responsible of nephrotoxicity in some patients. | ||
| + | The majority of NSAIDs inhibit competitively the initial dioxygenation (Rang & Dale, 2008; Ghosh et al., 2010). In general, these drugs block COX-1 in a quicker manner, whereas COX-2 inhibition is a more time-dependant event, and usually irreversible (Rang & Dale, 2008; Ghosh et al., 2010). The new COX-2 inhibitors exhibit PGHS-2 selectivity because they inhibit this isoform by a time-dependent, pseudoirreversible mechanism, whereas they inhibit PGHS-1 by a rapid, competitive, and reversible mechanism (Smith et al., 2000). | ||
| + | The inhibition mechanism consists of the entrance of the drug by the hydrophobic channel and the formation of hydrogen bonds with Arg120. This interaction prevents the fatty acids from entering the catalytic site. Selectivity of COX-2 inhibitors is mediated by the substitution of Ile523 in COX-1 with Val523 in COX-2, which results in the presence of a small side pocket adjacent to the active site channel, appreciably increasing the volume of the COX-2 active site (Rang & Dale, 2008; Ghosh et al., 2010; Smith et al., 2000). Acetylsalicylic acid makes its function by irreversible acetylation of COX-2 in Ser516 (Rang & Dale, 2008). | ||
| + | Last but not least, paracetamol, which do not interact neither with COX-1 nor with COX-2, may act as an analgesic and antipyretic drug by inhibition of COX-3 (Rang & Dale, 2008). | ||
| + | {| class="wikitable" | ||
| + | ! scope="col" | Pharmacologic group | ||
| + | ! scope="col" | Drug | ||
| + | |- | ||
| + | | Salicylates || Acetylsalicylic acid | ||
| + | |- | ||
| + | | rowspan="2" | Propionic | ||
| + | | Naproxen | ||
| + | |- | ||
| + | | Ibuprofen | ||
| + | |- | ||
| + | | Para-aminophenols || Paracetamol | ||
| + | |- | ||
| + | | Indolacetic || Indometacin | ||
| + | |- | ||
| + | | Pirrolacetic || Ketorolac | ||
| + | |- | ||
| + | | Phenilacetic || Diclofenac | ||
| + | |- | ||
| + | | Piranoidacetic || Etodolac | ||
| + | |- | ||
| + | | Anthranilic || Mefenamic acid | ||
| + | |- | ||
| + | | Nicotinic || Clonixin | ||
| + | |- | ||
| + | | Sulfonanilides || Nimesulide | ||
| + | |+ Table 1: Chemical variety of NSAIDs | ||
| + | |} | ||
| + | |||
{| border="1" style="margin: 1em auto 1em auto;" | {| border="1" style="margin: 1em auto 1em auto;" | ||
|- | |- | ||
| Line 87: | Line 125: | ||
|} | |} | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
| - | + | ||
==Reference== | ==Reference== | ||
Revision as of 16:39, 7 December 2010
COX-1 and COX-2, also called PGHS-1 and PGHS-2, regulate a key step in prostaglandins and thromboxanes synthesis and are the targets of nonsteroidal antiinflammatory drugs (NSAIDs) (Smith & Langenbach, 2001; Ghosh et al., 2010; Chandrasekharan et al., 2002). Prostaglandins are implicated in various pathophysiological processes such as inflammatory reaction, gastrointestinal cytoprotection, hemostasis and thrombosis, as well as renal hemodynamics (Smith & Langenbach, 2001; Ghosh et al., 2010; Smith et al., 2000). Whereas COX-1 presents a widespread constitutive expression, COX-2 is undetectable in most normal tissues (except for the central nervous system, kidneys, and seminal vesicles), but is induced by various inflammatory and mitogenic stimuli (Smith et al., 2000; Ghosh et al., 2010; Rang & Dale, 2008). More recently, a third isoform named COX-3 was identified as a COX-1 splicing variant. This new isoform may play a role in fever and pain processes (Ghosh et al., 2010; Chandrasekharan et al., 2002). Additionally, a high level of COX-2 expression is found usually in cancer cells (Ghosh et al., 2010). For example, COX-2 overexpression is related to poor-prognosis breast cancer (Boland et al., 2004; Barnes et al., 2007) and endometrial adenocarcinomas (Sales et al., 2008).
Contents |
About this structure (1,2)
PGHSs are bifunctional homodimers. Both COX-1 and COX-2 are membrane-bound enzymes and are present on the lumenal surfaces of the endoplasmic reticulum and of the inner and outer membranes of the nuclear envelope. However, recently, it has been demonstrated in cultured endothelial cells and fibroblasts that a fraction of COX-2 protein is localized to plasma membrane in caveolae-like structures (3). The primary structure of nascent COX-2 is of 604 amino acids and then it is processed into a mature form by removal of signal peptides giving a protein of 587 amino acids. PGHS-2 is variably glycosylated at two to four sites, leading to the formation of doublets or sometimes triplets on SDS-PAGE. Murine PGHS-2 peptide is presumed to be three times at Asn56, Asn130, and Asn396(NUEVA).
The COX monomer consists of three structural domains: the N-terminal EGF domain, a membrane binding domain (MBD) and a large C-terminal globular catalytic domain containing the heme binding site. The C-terminal segments beyond Pro583 (35 amino acids in COX-2) have not been resolved crystallographically. Collectively, these domains are made up of 25 alpha helices, seven 310 helices, four beta sheets as well as five disulfide bonds which contribute to the interface binding of the two individual monomers to complete the enzyme.
Protein domains
Epidermal Growth Factor Domain
The EGF and catalytic domains create the subunit interface in the dimer and place the two MBDs in a homodimer about 25 amstrongs apart. The EGF domains create a substantial portion of the dimer interface. EGF domains are common in several families of membrane proteins and secreted proteins. Typically, the EGF domain occurs at a position in the primary sequence N-terminal to a membrane anchor, such that these domains always occur on the extracytoplasmic face of the membrane. Some authors have suggested that the EGF domains may play a role in the insertion of COX into the lipid bilayer.
Membrane Binding Domain
PGHS-2 associate with only one face of the membrane bilayer through a monotopic membrane binding domain (MBD) that is comprised of four short, consecutive, amphipathic α-helices (helices A–D) that include residues 59-111 in human PGHS-2 (4). Three of the four helices lie roughly in the same plane while the last helix angles “upward” into the catalytic domain. Hydrophobic and aromatic residues protrude from these helices to create a hydrophobic surface that would interact with only one face of the lipid bilayer.
Catalytic Domain
The catalytic domain comprises the bulk of the COX monomer and is almost entirely composed of α-helical secondary structure. As said before COX are bifunctional proteins so we can discern two types of reactions: the heme-dependent bis-oxygenase or COX reaction that converts AA to PGG2 and the subsequent peroxidase (POX) reaction that reduces the 15-hydroperoxide of PGG2 to form PGH2.
Peroxidase Active Site Structure
The is in a large groove on the side opposite of the MBD. The structures of the peroxidase active sites of PGHSs are similar to those of other heme peroxidases. This site includes a heme group and the iron (III) in the center of this heme is coordinated by His-388 and by His-207.
Heme-dependent peroxidase activity is implicated in the formation of a proposed Tyr-385 radical, which is required for cyclooxygenase activity. Gln203 is also important in catalysis, although its function has not been resolved. Mutations of Gln203, His207, or His388 lead to a reduction or elimination of peroxidase activity.
The COXs bind 1 mole of ferric-protoporphyrin IX per mole monomer for full activity, as expected for a heme-dependent peroxidase.
Cyclooxygenase Active Site Structure
PGHS-1 and 2 monomers each contain a 25-°A hydrophobic channel that originates at the membrane binding domain and extends into the core of the globular domain. The MBD forms the mouth and the first half of the channel and allows arachidonate and O2 to enter directly from the apolar compartment of the lipid bilayer. Several amino acids composing the upper half of the channel are uniquely important in cyclooxygenase catalysis. Twenty-four residues line the hydrophobic cyclooxygenase active site with only one difference between the isozymes—Ile at position 523 in PGHS-1 and Val at position 523 in PGHS-2. Amino acids lining the hydrophobic cyclooxygenase active site channel include Leu117, Arg120, Phe205, Phe209, Val344, Ile345, Tyr348, Val349, Leu352, Ser353, Tyr355, Leu359, Phe381, Leu384, Tyr385, Trp387, Phe518, Ile/Val523, Gly526, Ala527, Ser530, Leu531, Gly533, Leu534. Only three of the channel residues are polar (Arg120, Ser353, and Ser530). Tyr 385 in its radical form is the responsible for abstracting a proton from arachidonic acid during its conversion to PGG2. Arg120, which is positioned about midway between the mouth and the apex of the active site (7), binds to the carboxylate groups of fatty acids and many NSAIDs.
|
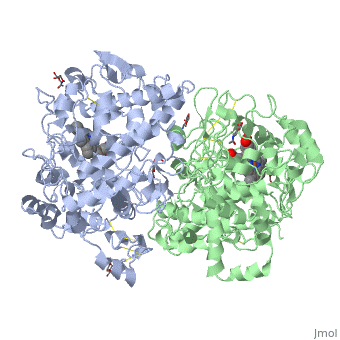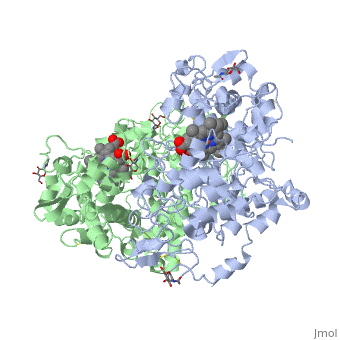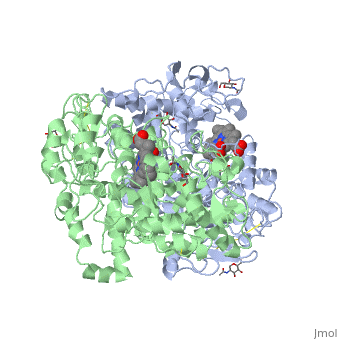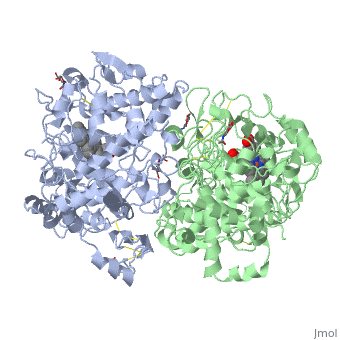
Function
COX-2, unlike COX-1, is induced in inflammatory cells when they are activated by various inflammatory and mitogenic stimuli [1]. Its function is to produce the prostanoid mediators of the inflammation, although there are some significant exceptions. For example, there is a considerable pool of “constitutive” COX-2 present in the central nervous system (CNS) and some other tissues, although its function is not yet completely clear [2]. Moreover, COX-1, that is present in most tissues, has a “housekeeping” role in the body, being involved in tissue homeostasis, and is responsible for the production of prostaglandins involved in gastric cytoprotection, platelet aggregation, renal blood flow autoregulation and the initiation of parturition [2].
Physiological Regulation
COX-2 overexpression is very important because it has significant tissue-specific consequences and is associated with inflammatory diseases, cancers and term/preterm labour, thus making COX-2 an important target for pharmacological intervention [1]. It is important to know that the expression of COX-2 in many specialized cell types appears to be differentially sensitive to the different stimuli that regulate the unique physiological activities of each tissue [13]. It is known that the physiological regulation can be produced at various levels [1]: - Transcriptional regulation - Post-transcriptional regulation of COX-2 via its 3’-UTR - COX-2 3’-UTRs: miRNAs (microRNAs) and alternative polyadenylation
Transcriptional regulation [1]
Transcriptional activation of COX-2 occurs quickly and transiently in response to different stimuli, for example: pathogens, cytokines, nitric oxide, irradiation, growth factors and various extracellular ligands. The 5-UTR (untranslated region) of the COX-2 gene has several transcription factor response elements, including two NF-κB (nuclear factor κB) motifs, two AP-1 (activator protein 1) sites and two CREs (cAMP-response elements), among others [2]. Transcriptional regulation of COX-2 may also be physically influenced by chromatin remodelling events such as changes in acetylation status of histones and non-histone proteins. For example, the acetylation of NF-κB components by the transcriptional coativator p300 (histone acetyltransferase [HAT]) can activate the COX-2 expression[3], while the hypermethylation of the CpG islands results in transcriptional silencing [4]. It is also known that the histone deacetylase inhibitors (iHDAC) suppress the activation of the expression in human primary myometrtial cells [5] and in cancer cell lines [6], by preventing the binding of the transcription factor, c-Jun, to the COX-2 promoter [6].
Post-transcriptional regulation [1]
Via 3’-UTR
The 3’-UTR of COX-2 is a complex region that contains multiple copies of AREs (AU-rich elements) throughout sequence, which, when bound by specific trans-acting ARE-binding factors, influence COX-2 mRNA stability and also translational efficiency [7]. A lot of studies have introduced a new model to the gene regulation of COX-2 by investigating the combined contribution of both transcription and mRNA stability events. For example, one group has reported that the binding of the protein CUGBP2 (CUG triplet repeat, RNA-binding protein 2) in specific AREs within the first 60 nucleotides of the COX-2 3’-UTR can stabilize the COX-2 mRNA inhibiting its translation [8]. Also, there is evidence that mitogenic inhibitors (e.g. taxanes) can control COX-2 transcription via PKC (Protein Kinase C)-p38 MAPK (Mitogen-Activated Protein Kinase) signaling cascade and it is known that the stability of COX-2 mRNA can be controlled by the binding of HuR (a mRNA-stabilizing factor) to AREs in 3’-UTR of COX-2.
miRNAs (microRNAs)
A recent study has demonstrated that the microRNAs miR-101a and miRNA-199a can interact with the COX-2 3’-UTR in vitro repressing its translation [10].
Alternative polyadenylation
The human COX-2 3’-UTR has several polyadenylation sites. COX-2 uses two alternative polyadenylation sites, in a tissue-specific manner, which derives in the formation of 2 COX-2 mRNAs: one with 2.8 kb and another one with 4.6 kb [11]. It is known that selection of the proximal polyadenylation signal is enhanced by presence of additional USEs (Upstream Sequence Elements) where four RNA-binding proteins (U1A, PTB, p54nrb and PSF) can bind, enhancing the recruitment and stabilization of core adenylation factors on the COX-2 mRNA [12].
NSAIDs
Non-steroid anti-inflammatory drugs are a chemically heterogeneous group of compounds whose major function is the inhibition of cyclooxygenases (Table 1). Apart from their anti-inflammatory effect, they also present analgesic and antipyretic properties (Rang & Dale, 2008). Classical NSAIDs, as salicylate or phenoprofen, are mostly inhibitors of both isoenzymes, although each isoform is inhibited in a different level. Chronic users of NSAIDs develop gastric ulcers or gastrointestinal complications, explained by the inhibition of COX-1. For this reason, selective inhibitors of COX-2, as celecoxib, valdecoxib and etoricoxib, have been developed (Ghosh et al., 2010, Rang & Dale, 2008). They don’t cause gastric pathology, but it has been proved to be responsible of nephrotoxicity in some patients. The majority of NSAIDs inhibit competitively the initial dioxygenation (Rang & Dale, 2008; Ghosh et al., 2010). In general, these drugs block COX-1 in a quicker manner, whereas COX-2 inhibition is a more time-dependant event, and usually irreversible (Rang & Dale, 2008; Ghosh et al., 2010). The new COX-2 inhibitors exhibit PGHS-2 selectivity because they inhibit this isoform by a time-dependent, pseudoirreversible mechanism, whereas they inhibit PGHS-1 by a rapid, competitive, and reversible mechanism (Smith et al., 2000). The inhibition mechanism consists of the entrance of the drug by the hydrophobic channel and the formation of hydrogen bonds with Arg120. This interaction prevents the fatty acids from entering the catalytic site. Selectivity of COX-2 inhibitors is mediated by the substitution of Ile523 in COX-1 with Val523 in COX-2, which results in the presence of a small side pocket adjacent to the active site channel, appreciably increasing the volume of the COX-2 active site (Rang & Dale, 2008; Ghosh et al., 2010; Smith et al., 2000). Acetylsalicylic acid makes its function by irreversible acetylation of COX-2 in Ser516 (Rang & Dale, 2008). Last but not least, paracetamol, which do not interact neither with COX-1 nor with COX-2, may act as an analgesic and antipyretic drug by inhibition of COX-3 (Rang & Dale, 2008).
| Pharmacologic group | Drug |
|---|---|
| Salicylates | Acetylsalicylic acid |
| Propionic | Naproxen |
| Ibuprofen | |
| Para-aminophenols | Paracetamol |
| Indolacetic | Indometacin |
| Pirrolacetic | Ketorolac |
| Phenilacetic | Diclofenac |
| Piranoidacetic | Etodolac |
| Anthranilic | Mefenamic acid |
| Nicotinic | Clonixin |
| Sulfonanilides | Nimesulide |
| Drug | Coefficient of selectivity (IC50Cox-1/IC50Cox-2) |
|---|---|
| Ketorolac | |
| Naproxen | |
| Ibuprofen | |
| Indometacin | |
| Acetylsalicylic acid | |
| Diclofenac | |
| Valdecoxib | |
| Etoricoxib | |
Reference
- Ghosh N, Chaki R, Mandal V, Mandal SC. COX-2 as a target for cancer chemotherapy. Pharmacol Rep. 2010 Mar-Apr;62(2):233-44. PMID:20508278
- Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145-82. PMID:10966456 doi:10.1146/annurev.biochem.69.1.145
- Rang HP, Dale MM, Ritter JM, Flower RJ. 2008. Pharmacology. Elsevier. 6th edition. 844 p.
- Smith WL, Langenbach R. Why there are two cyclooxygenase isozymes. J Clin Invest. 2001 Jun;107(12):1491-5. PMID:11413152 doi:10.1172/JCI13271
- Chandrasekharan NV, Dai H, Roos KL, Evanson NK, Tomsik J, Elton TS, Simmons DL. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc Natl Acad Sci U S A. 2002 Oct 15;99(21):13926-31. Epub 2002 Sep 19. PMID:12242329 doi:10.1073/pnas.162468699
- Garavito RM, Mulichak AM. The structure of mammalian cyclooxygenases. Annu Rev Biophys Biomol Struct. 2003;32:183-206. Epub 2003 Feb 5. PMID:12574066 doi:10.1146/annurev.biophys.32.110601.141906
- Perrone G, Zagami M, Altomare V, Battista C, Morini S, Rabitti C. COX-2 localization within plasma membrane caveolae-like structures in human lobular intraepithelial neoplasia of the breast. Virchows Arch. 2007 Dec;451(6):1039-45. Epub 2007 Sep 13. PMID:17851687 doi:10.1007/s00428-007-0506-4
- Spencer AG, Thuresson E, Otto JC, Song I, Smith T, DeWitt DL, Garavito RM, Smith WL. The membrane binding domains of prostaglandin endoperoxide H synthases 1 and 2. Peptide mapping and mutational analysis. J Biol Chem. 1999 Nov 12;274(46):32936-42. PMID:10551860
- Luong C, Miller A, Barnett J, Chow J, Ramesha C, Browner MF. Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nat Struct Biol. 1996 Nov;3(11):927-33. PMID:8901870
- Kurumbail RG, Stevens AM, Gierse JK, McDonald JJ, Stegeman RA, Pak JY, Gildehaus D, Miyashiro JM, Penning TD, Seibert K, Isakson PC, Stallings WC. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 1996 Dec 19-26;384(6610):644-8. PMID:8967954 doi:http://dx.doi.org/10.1038/384644a0
Proteopedia Page Contributors and Editors (what is this?)
Cristina Murga, Michal Harel, David Canner, María Laura Saiz Álvarez, Alexander Berchansky
