Structural linkage between ligand discrimination and receptor activation by type I interferons
Christoph Thomas, Ignacio Moraga, Doron Levin, Peter O. Krutzik, Yulia Podoplelova, Angelica Trejo, Choongho Lee, Ganit Yarden, Susan E. Vleck, Jeffrey S. Glenn, Garry P. Nolan, Jacob Piehler, Gideon Schreiber, K. Christopher Garcia[1]
Molecular Tour
IFNs were the first cytokines discovered more than half a century ago as agents that interfere with viral infection. Since then, IFNs have been established as pleiotropic, multifunctional proteins in the early immune response, exhibiting pronounced antiproliferative effects on cells, in addition to their strong immunomodulatory and antiviral activity. Due to their potency and diverse biological activities, IFNs are used for the treatment of several human diseases, including hepatitis C, multiple sclerosis and certain types of cancer.
All initiate signaling by binding to the same receptor composed of two subunits called and . The intracellular domains (ICDs) of IFNAR1 and IFNAR2 are associated with the Janus kinases (Jaks) Tyk2 and Jak1, respectively. Upon ligand binding by the IFNAR chains and formation of the signaling complex, these tyrosine kinases trans-phosphorylate and thereby activate each other. Subsequently, the activated Jaks phosphorylate STAT1, STAT2 and STAT3, which translocate into the nucleus and activate the transcription of hundreds of IFN-stimulated genes. To gain insight into how type I IFNs engage their receptor chains, how the receptor system is able to recognize the large number of different ligands, and how different IFN ligands can evoke different physiological activities, we determined the crystal structures of unliganded , the binary complex , and the ternary ligand-receptor complexes of and binding both receptor chains. A final theoretical ternary structure including was also created. These structures, in conjunction with biochemical and cellular experiments, reveal that the type I IFN receptor uses a mode of ligand interaction that is unique among cytokine receptors, but conserved between different IFNs.
Interactions Between IFNAR & IFN
IFNAR2-IFN interaction
interacts primarily with the . Arg33(IFN) appears to be the for the interaction of the IFN ligand with IFNAR2. It forms an with the main chain carbonyl oxygen atoms of . This residue is present in IFNα, IFNω, IFNβ and IFNε. Two hydrophobic interaction clusters are present in the interface: is formed between Leu15 and Met16 of the IFN molecule and Trp100 and Ile103 of IFNAR2; comprises Leu26, Phe27, Leu30 and Val142 of the ligand and Met46, Leu52, Val80 and the methyl group of Thr44 of the receptor. Replacing reduces affinity by three orders of magnitude (the second most important residue for binding). This is surprising, as it is . One reason for its importance might be a .
Most of the residues involved in the IFNα2-IFNAR2 interaction are also found in the IFNω-IFNAR2 interface of the IFNω ternary complex.

A significant difference in the IFNAR2 interface between and IFNw is related to , which is replaced with Lys152 in . In the , this residue forms an with Glu147(IFN), but . The IFNa-IFNAR2 interface buries ~1841 Å^2 of surface area.
IFNAR1-IFN interaction
Because of the lower resolution of the IFNα ternary complex, we focused on the in our analysis of the IFN-IFNAR1 interface (Figure 4). However, the IFNω ternary complex on the IFNα2 ternary complex, suggesting similar binding. In the , the only contains one hotspot residue we could experimentally confirm, . Substituting this residue by alanine reduces the affinity to all tested IFN ligands by more than 20-fold (Table 1). The interaction map for the IFNω/IFNAR1 interface suggests that . However, the F238A mutant of IFNAR1 could not be produced with sufficient yields for quantitative binding studies. On , mutation studies have shown that a charge-reversal (Arg 120 on IFNα) lead to a total loss of activity.
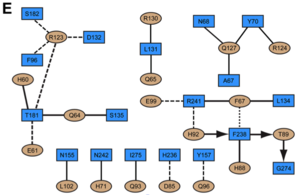
Indeed, this residue forms a in addition to (Figure 4B and 4E). Substitution of glutamate for Arg123IFN would lead to electrostatic repulsion with Asp132R1. The low affinity of IFNAR1 for the ligand appears to be functionally relevant, as weak binding to IFNAR1 is conserved between all alpha IFNs. Three amino acid substitutions on IFNα2 at positions His57, Glu58 and Ser61 to AAA or to YNS confer tighter binding to IFNAR1, but leave the affinity to IFNAR2 essentially unaltered.
Implications for the binding mode of IFNβ
30% and 33% sequence identity with and IFNα2, respectively. in our ternary complex structure leads of side chains ( and ) with the receptors, indicating that the IFNβ ligand could be easily accommodated by the receptors in a position similar to IFNω and IFNα2. Furthermore, the shows that . As a result, Ala19IFN, when mutated to tryptophan, promotes an increased binding affinity to IFNAR2, which is a result of the (as shown by double mutant cycle analysis).
Structural Movements
Structural pertubations upon binding
One of the more controversial aspects of cytokine signalling is whether receptor binding is sufficient to generate activity, or it has to be accompanied by structural pertubations. The type I interferon complex is one of the only cytokine receptor complexes were the structures of all the components making up the biologically active complex were determined to high resolution in their free and bound forms. of the unbound NMR structure with the ternary complex structure of interferon shows a small expansion during complex formation. (Figure S3).
Conversely, both in IFNAR1 and IFNAR2 undergo major domain movements upon binding. Using the D1 domain as anchor, a of IFNAR2 upon binding, on a scale of 6-12 Å is observed (comparison of the unbound receptor (1n6u) with the binary IFNα2-IFNAR2 complex). However, also the superimposition of the IFNα2-IFNAR2 binary complex onto IFN-IFNAR2 in the ternary complexes of 6-9 Å, and even between the ternary IFNα and IFNω complexes a movement of 3-5 Å is observed (Figure S3). As D2 is engaged in crystal contacts in all three structures, the large variations in D2 may suggest some flexibility in the hinge of D1 and D2 in IFNAR2. Still, these movements could change the proximity/orientation of the ICDs and associated Jaks within the cell.
The low affinity binding receptor, IFNAR1 also upon ternary complex formation (Figure S3). When using D1 as anchor, D3 is moving inwards (closing upon interferon) by ~15 Å. This would generate an even larger movement of the transmembrane proximal D4 domain and the transmembrane helix. The IFNAR1 conformation is very similar when ligated to IFNα2 or IFNω, and is not supported by crystal contacts. Contrary to D3, D4 seems to be highly flexible (even more than D2 of IFNAR2). Interestingly, the only hotspot residue on IFNα2 (Arg 120) as well as Leu 117 make contacts with Tyr 70 on D1 of IFNAR1, which is one of the most important residues on the interferon binding site on IFNAR1. This may suggest that D1 makes a futile encounter complex with interferon, which may develop into a signaling complex upon structural rearrangement of the receptor. This would result in a very signification inwards movement of the intracellular, unstructured domain of IFNAR1, promoting proximity between Jak1 and Tyk2 that is required for reciprocal trans-phosphorylation and phosphorylation of specific tyrosine residues in the intracellular domains of IFNAR1 and IFNAR2. Moreover, one may suggest that the conformational change by itself will be responsible for a reduced binding affinity of IFNAR1 and may slow down the rate of ligand association to IFNAR1 directly from solution. The here proposed mechanism would result in a much tighter control on interferon signalling, as random events of receptor proximity will not be able to overcome the activation energy needed for receptor structural rearrangements, which require specific ligand binding. The overall mechanism of activation may be even more complex, if indeed the D2 domain of IFNAR2 is also moving upon ternary complex formation (as suggested by the structures). In this case, a would be required to form a fruitful reaction complex (Figure S3).




