User:Yunlong Zhao/Sandbox 1
From Proteopedia
(Difference between revisions)
| Line 15: | Line 15: | ||
== The allosteric mechanism of heparin-based anticoagulant activation == | == The allosteric mechanism of heparin-based anticoagulant activation == | ||
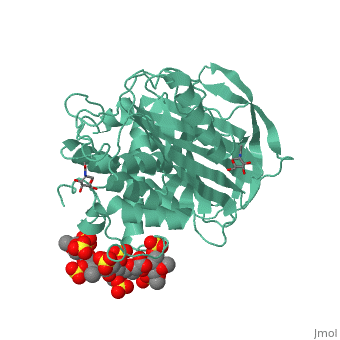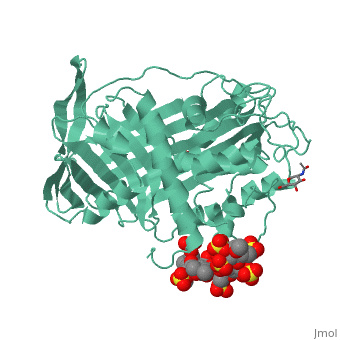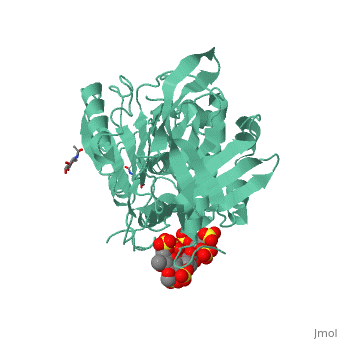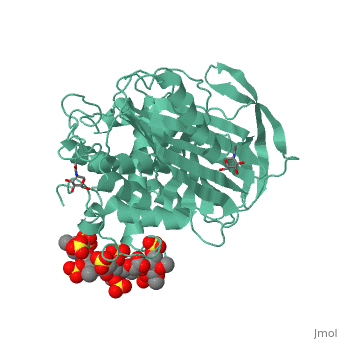
| - | A model of anticoagulant activation of antithrombin by heparin was described as an allosteric mechanism. In another word, heparin chain binds to antithrombin and stabilizes the inhibitory conformation (the inhibitory loop is outward) or alters the antithrombin conformation to facilitate the antithrombin-protease interaction. Early published crystal structure of pentasaccharide-bond antithrombin provided many details of the interaction and validated this hypothesis to some extents. Pentasaccharide binds to the <scene name='58/584311/Binding_helices/3'>helical region</scene> distal to the inhibitory loop. | + | A model of anticoagulant activation of antithrombin by heparin was described as an allosteric mechanism. In another word, heparin chain binds to antithrombin and stabilizes the inhibitory conformation (the inhibitory loop is outward) or alters the antithrombin conformation to facilitate the antithrombin-protease interaction. Early published crystal structure of pentasaccharide-bond antithrombin provided many details of the interaction and validated this hypothesis to some extents. Pentasaccharide binds to the <scene name='58/584311/Binding_helices/3'>helical region</scene> distal to the inhibitory loop, which is pushed outward to make the contact with protease (eg. thrombin or factor Xa) easier. Although the plenty of charge on heparin strongly suggests that majority of interaction between antithrombin and heparin in solution is the electrostatic force, the distance between those two appears to be very close when heparin approaches AT-III during crystallization. This short distance implies that multiple hydrogen bond could be formed on the interface with helical region, supported by the <scene name='58/584311/Hydrogen_bond/2'>crystal structure</scene>. The residues from the helical region of heparin-binding domain that contribute to H-bonding interaction include Arg129, Lys125, Lys 11, Lys114, Arg13, Asn45, Arg46, Arg47 and Glu113 (shown in lime). Most of those residues are positively charged and can potentially attract the negative charges on sulfate groups of heparin to initialize those sidechains orientation. Interestingly, if comparing with the non-heparin structure of AT-III, some conformation differences are observed in pentasaccharide-binding structure. For example, the attachment of pentasaccharide to the helical region induces a unique helix extension (shown in blue). Three positively charged sidechains Lys133, Lys136 and Arg136 on the extended helix are most possible to be affected by the pentasaccharide-bindng. Those secondary structures rearrangement may offer some torsion forces and finally lead the inhibitory loop to shift allosterically. |
| - | Green scene 2 will be the the interface structure plus heparin | ||
| - | |||
| - | This is a sample scene created with SAT to <scene name="/12/3456/Sample/1">color</scene> by Group, and another to make <scene name="/12/3456/Sample/2">a transparent representation</scene> of the protein. You can make your own scenes on SAT starting from scratch or loading and editing one of these sample scenes. | ||
| - | |||
| - | </StructureSection> | ||
== References == | == References == | ||
<references/> | <references/> | ||
Revision as of 13:38, 11 May 2014
Anticoagulation effects of heparin and the interaction with antithrombin
| |||||||||||