This old version of Proteopedia is provided for student assignments while the new version is undergoing repairs. Content and edits done in this old version of Proteopedia after March 1, 2026 will eventually be lost when it is retired in about June of 2026.
Apply for new accounts at the new Proteopedia. Your logins will work in both the old and new versions.
2gk1
From Proteopedia
(Difference between revisions)
| Line 1: | Line 1: | ||
| - | + | ==X-ray crystal structure of NGT-bound HexA== | |
| - | === | + | <StructureSection load='2gk1' size='340' side='right' caption='[[2gk1]], [[Resolution|resolution]] 3.25Å' scene=''> |
| - | + | == Structural highlights == | |
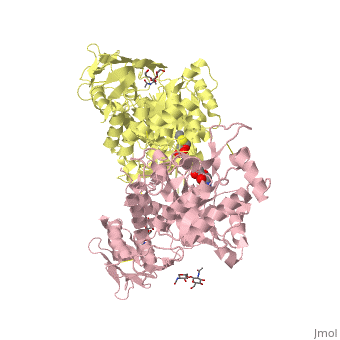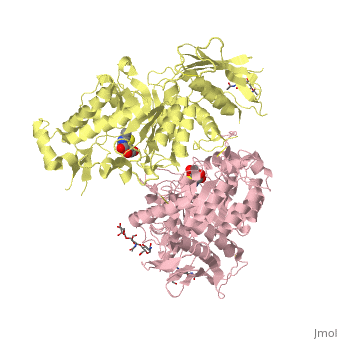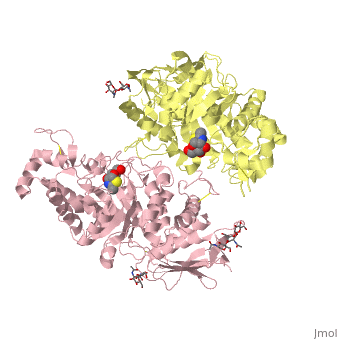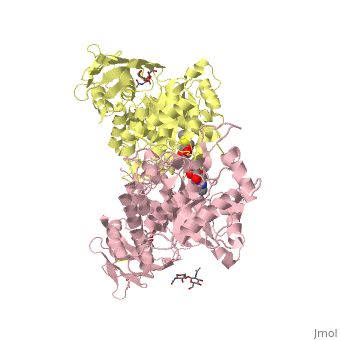
| + | <table><tr><td colspan='2'>[[2gk1]] is a 8 chain structure with sequence from [http://en.wikipedia.org/wiki/Homo_sapiens Homo sapiens]. Full crystallographic information is available from [http://oca.weizmann.ac.il/oca-bin/ocashort?id=2GK1 OCA]. For a <b>guided tour on the structure components</b> use [http://oca.weizmann.ac.il/oca-docs/fgij/fg.htm?mol=2GK1 FirstGlance]. <br> | ||
| + | </td></tr><tr><td class="sblockLbl"><b>[[Ligand|Ligands:]]</b></td><td class="sblockDat"><scene name='pdbligand=BMA:BETA-D-MANNOSE'>BMA</scene>, <scene name='pdbligand=NGT:3AR,5R,6S,7R,7AR-5-HYDROXYMETHYL-2-METHYL-5,6,7,7A-TETRAHYDRO-3AH-PYRANO[3,2-D]THIAZOLE-6,7-DIOL'>NGT</scene>, <scene name='pdbligand=NAG:N-ACETYL-D-GLUCOSAMINE'>NAG</scene><br> | ||
| + | <tr><td class="sblockLbl"><b>[[Related_structure|Related:]]</b></td><td class="sblockDat">[[2gjx|2gjx]]</td></tr> | ||
| + | <tr><td class="sblockLbl"><b>Activity:</b></td><td class="sblockDat"><span class='plainlinks'>[http://en.wikipedia.org/wiki/Beta-N-acetylhexosaminidase Beta-N-acetylhexosaminidase], with EC number [http://www.brenda-enzymes.info/php/result_flat.php4?ecno=3.2.1.52 3.2.1.52] </span></td></tr> | ||
| + | <tr><td class="sblockLbl"><b>Resources:</b></td><td class="sblockDat"><span class='plainlinks'>[http://oca.weizmann.ac.il/oca-docs/fgij/fg.htm?mol=2gk1 FirstGlance], [http://oca.weizmann.ac.il/oca-bin/ocaids?id=2gk1 OCA], [http://www.rcsb.org/pdb/explore.do?structureId=2gk1 RCSB], [http://www.ebi.ac.uk/pdbsum/2gk1 PDBsum]</span></td></tr> | ||
| + | <table> | ||
| + | == Disease == | ||
| + | [[http://www.uniprot.org/uniprot/HEXA_HUMAN HEXA_HUMAN]] Defects in HEXA are the cause of GM2-gangliosidosis type 1 (GM2G1) [MIM:[http://omim.org/entry/272800 272800]]; also known as Tay-Sachs disease. GM2-gangliosidosis is an autosomal recessive lysosomal storage disease marked by the accumulation of GM2 gangliosides in the neuronal cells. GM2G1 is characterized by GM2 gangliosides accumulation in the absence of HEXA activity, leading to neurodegeneration and, in the infantile form, death in early childhood. GM2G1 has an increased incidence among Ashkenazi Jews and French Canadians in eastern Quebec. It exists in several forms: infantile (most common and most severe), juvenile and adult (late onset).<ref>PMID:2970528</ref> <ref>PMID:2522679</ref> <ref>PMID:2144098</ref> <ref>PMID:1837283</ref> <ref>PMID:1532289</ref> <ref>PMID:1302612</ref> <ref>PMID:1301189</ref> <ref>PMID:1301190</ref> <ref>PMID:8490625</ref> <ref>PMID:8445615</ref> <ref>PMID:7951261</ref> <ref>PMID:7837766</ref> <ref>PMID:7717398</ref> <ref>PMID:8581357</ref> <ref>PMID:7898712</ref> <ref>PMID:8757036</ref> <ref>PMID:9150157</ref> <ref>PMID:9338583</ref> <ref>PMID:9375850</ref> <ref>PMID:9401008</ref> <ref>PMID:9603435</ref> <ref>PMID:14566483</ref> [[http://www.uniprot.org/uniprot/HEXB_HUMAN HEXB_HUMAN]] Defects in HEXB are the cause of GM2-gangliosidosis type 2 (GM2G2) [MIM:[http://omim.org/entry/268800 268800]]; also known as Sandhoff disease. GM2-gangliosidosis is an autosomal recessive lysosomal storage disease marked by the accumulation of GM2 gangliosides in the neuronal cells. GM2G2 is clinically indistinguishable from GM2-gangliosidosis type 1, presenting startle reactions, early blindness, progressive motor and mental deterioration, macrocephaly and cherry-red spots on the macula.<ref>PMID:1720305</ref> <ref>PMID:1531140</ref> <ref>PMID:8357844</ref> <ref>PMID:7626071</ref> <ref>PMID:7557963</ref> <ref>PMID:7633435</ref> <ref>PMID:8950198</ref> <ref>PMID:9401004</ref> <ref>PMID:9856491</ref> <ref>PMID:9694901</ref> | ||
| + | == Function == | ||
| + | [[http://www.uniprot.org/uniprot/HEXA_HUMAN HEXA_HUMAN]] Responsible for the degradation of GM2 gangliosides, and a variety of other molecules containing terminal N-acetyl hexosamines, in the brain and other tissues. The form B is active against certain oligosaccharides. The form S has no measurable activity. [[http://www.uniprot.org/uniprot/HEXB_HUMAN HEXB_HUMAN]] Responsible for the degradation of GM2 gangliosides, and a variety of other molecules containing terminal N-acetyl hexosamines, in the brain and other tissues. | ||
| + | == Evolutionary Conservation == | ||
| + | [[Image:Consurf_key_small.gif|200px|right]] | ||
| + | Check<jmol> | ||
| + | <jmolCheckbox> | ||
| + | <scriptWhenChecked>select protein; define ~consurf_to_do selected; consurf_initial_scene = true; script "/wiki/ConSurf/gk/2gk1_consurf.spt"</scriptWhenChecked> | ||
| + | <scriptWhenUnchecked>script /wiki/extensions/Proteopedia/spt/initialview01.spt</scriptWhenUnchecked> | ||
| + | <text>to colour the structure by Evolutionary Conservation</text> | ||
| + | </jmolCheckbox> | ||
| + | </jmol>, as determined by [http://consurfdb.tau.ac.il/ ConSurfDB]. You may read the [[Conservation%2C_Evolutionary|explanation]] of the method and the full data available from [http://bental.tau.ac.il/new_ConSurfDB/chain_selection.php?pdb_ID=2ata ConSurf]. | ||
| + | <div style="clear:both"></div> | ||
| + | <div style="background-color:#fffaf0;"> | ||
| + | == Publication Abstract from PubMed == | ||
| + | Lysosomal beta-hexosaminidase A (Hex A) is essential for the degradation of GM2 gangliosides in the central and peripheral nervous system. Accumulation of GM2 leads to severely debilitating neurodegeneration associated with Tay-Sachs disease (TSD), Sandoff disease (SD) and AB variant. Here, we present the X-ray crystallographic structure of Hex A to 2.8 A resolution and the structure of Hex A in complex with NAG-thiazoline, (NGT) to 3.25 A resolution. NGT, a mechanism-based inhibitor, has been shown to act as a chemical chaperone that, to some extent, prevents misfolding of a Hex A mutant associated with adult onset Tay Sachs disease and, as a result, increases the residual activity of Hex A to a level above the critical threshold for disease. The crystal structure of Hex A reveals an alphabeta heterodimer, with each subunit having a functional active site. Only the alpha-subunit active site can hydrolyze GM2 gangliosides due to a flexible loop structure that is removed post-translationally from beta, and to the presence of alphaAsn423 and alphaArg424. The loop structure is involved in binding the GM2 activator protein, while alphaArg424 is critical for binding the carboxylate group of the N-acetyl-neuraminic acid residue of GM2. The beta-subunit lacks these key residues and has betaAsp452 and betaLeu453 in their place; the beta-subunit therefore cleaves only neutral substrates efficiently. Mutations in the alpha-subunit, associated with TSD, and those in the beta-subunit, associated with SD are discussed. The effect of NGT binding in the active site of a mutant Hex A and its effect on protein function is discussed. | ||
| - | + | Crystallographic structure of human beta-hexosaminidase A: interpretation of Tay-Sachs mutations and loss of GM2 ganglioside hydrolysis.,Lemieux MJ, Mark BL, Cherney MM, Withers SG, Mahuran DJ, James MN J Mol Biol. 2006 Jun 16;359(4):913-29. Epub 2006 Apr 27. PMID:16698036<ref>PMID:16698036</ref> | |
| - | + | ||
| - | + | From MEDLINE®/PubMed®, a database of the U.S. National Library of Medicine.<br> | |
| - | + | </div> | |
| - | + | ||
| - | + | ||
| - | + | ||
==See Also== | ==See Also== | ||
*[[Beta-Hexosaminidase|Beta-Hexosaminidase]] | *[[Beta-Hexosaminidase|Beta-Hexosaminidase]] | ||
| - | + | == References == | |
| - | == | + | <references/> |
| - | + | __TOC__ | |
| + | </StructureSection> | ||
[[Category: Beta-N-acetylhexosaminidase]] | [[Category: Beta-N-acetylhexosaminidase]] | ||
[[Category: Homo sapiens]] | [[Category: Homo sapiens]] | ||
Revision as of 08:46, 30 September 2014
X-ray crystal structure of NGT-bound HexA
| |||||||||||