AChE substrate
Dear readers, this page presents only a small part of the great world of the acetylcholinesterase inhibitors. So, please see also our pages AChE inhibitors and substrates (Part II), AChE inhibitors and substrates (Part III), AChE bivalent inhibitors and AChE bivalent inhibitors (Part II). Solution of the three-dimensional (3D) structure
of Torpedo californica acetylcholinesterase (TcAChE)
in 1991 [1] opened up new horizons in research on an enzyme that had already been the subject of intensive investigation. The unanticipated structure of this extremely rapid enzyme, in which the active site was found to be buried at the bottom of a , lined by (colored dark magenta), led to a revision of the views then held concerning substrate traffic, recognition and hydrolysis [2]. This led to a series of theoretical and experimental studies, which took advantage of recent advances in theoretical techniques for treatment of proteins, such as
molecular dynamics and electrostatics and to site-directed mutagenesis, utilizing suitable expression
systems. Acetylcholinesterase hydrolysizes the neurotransmitter acetylcholine , producing group. ACh directly binds (via its nucleophilic Oγ atom) within the (ACh/TcAChE structure 2ace). The residues are also important in the ligand recognition [3]. After this binding acetylcholinesterase ACh.
AChE monovalent inhibitors
Organophosphorus acid anhydride nerve agents
Organophosphorus (OP) acid anhydride nerve agents are potent inhibitors which rapidly phosphonylate AChE and then may undergo an internal dealkylation reaction (called "aging") to produce an OP-enzyme conjugate that cannot be reactivated.
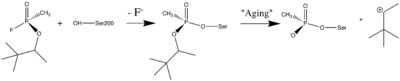
Reaction between Ser200Oγ and Soman, assuming an in-line attack by the Oγ, followed by spontaneous dealkylation of the O-pinacolyl group.
Soman
As was mentioned above, AChE hydrolysizes the neurotransmitter , producing group. directly binds catalytic (via its nucleophilic Oγ atom). , O-(1,2,2-trimethylpropyl) methylphosphonofluoridate (fluorine atom is colored violet and phosphorus atom is colored darkmagenta), is one of the most toxic OPs. Soman inhibits AChE by to catalytic Ser200, . This process implicates nucleophilic attack of the Ser200 nucleophilic Oγ atom on the phosphorus atom of soman, with concomitant departure of its fluoride atom. After that AChE catalyzes the of the soman or other OP. This causes irreversible inhibition of AChE, "aged" soman/AChE conjugate can not be reactivated. However, before “aging”, at the step of , AChE can be by nucleophiles, such as pralidoxime (2-PAM), resulting in of the phosphonyl adduct from Ser200 Oγ.
At the (2wfz) the catalytic His440 forms hydrogen bonds with Ser200 Oγ and Glu327 Oε1 via its Nε2 and Nδ1 nitrogens, respectively. The O2 atom of soman is within hydrogen bonding distance of His440 Nε2. Soman O1 mimicks carbonyl oxygen of ACh. A water molecule 1001 interacting with soman O2 is represented as a red ball. The active site residues of the nonaged soman/TcAChE are colored yellow. The O2 atom of the (2wg0) forms a salt bridge with His440 Nε2. The active site residues of the aged soman/TcAChE are colored pink. of the structures of the nonaged (2wfz) and aged (2wg0) conjugates reveals a small, but important, change within the active site - the imidazole ring of His440 is tilted back to a native-like conformation after dealkylation. The water molecule 1001, which interacts with soman O2 in the nonaged crystal structure, is not within hydrogen bonding distance of O2 in the aged crystal structure. 2-PAM binds poorly to the nonaged phosphonylated enzyme (its electron density was not found) and binds in an after soman aging to TcAChE (2wg1) [4].
To understand the basis for irreversible inhibition, the obtained by reaction of TcAChE with soman was solved by X-ray crystallography to 2.2Å resolution (1som). The highest positive difference density peak corresponded to the OP phosphorus and was located within covalent bonding distance of the active-site serine (S200). The are within hydrogen-bonding distance of four potential donors from catalytic subsites of the enzyme, suggesting that electrostatic forces significantly stabilize the aged enzyme. The methyl group of soman occupies the , bounded by Trp233, Phe288, and Phe290 [5].
Sarin
Sarin, O-isopropylmethylphosponofluoridate, is an other toxic OP compound. It is also inhibits AChE by covalent binding to the catalytic Ser200. The active sites of aged (1cfj) and aged soman-TcAChE (1som and 2wg0) are almost identical and provided structural models for the negatively charged, tetrahedral intermediate that occurs during deacylation with the ACh.
There are four hydrogen bond donors (red dotted lines) to the anionic phosphonyl oxygen atoms: the backbone amide nitrogen atoms of Ala201, Gly118, and Gly119, as well as His440 Nε2. The sarin methyl carbon (colored cyan) is within non-bonded contact distances (black dotted lines) of Phe288 and Phe290 in the acyl binding pocket [5].
DFP
, diisopropylphosphorofluoridate, is an other toxic OP nerve agent. It is also inhibits AChE by covalent binding to the catalytic Ser200. As in the case with soman (1som) and sarin (1cfj), there are four hydrogen bond donors (dotted lines) to the anionic phosphonyl oxygen atoms: the backbone amide nitrogen atoms of Ala201, Gly118, and Gly119, as well as His440 Nε2 at the of aged DFP-TcAChE (2dfp). Phosphorylation with DFP caused an unexpected distortion in the main chain of a loop that includes residues F288 and F290 of the TcAChE acyl binding pocket. F288 and F290 move significantly in the DFP-TcAChE structure (green), in comparison to their positions in the native enzyme (2ace). This is the first major conformational change reported in the active site of any AChE−ligand complex, and it offers a structural explanation for the substrate selectivity of AChE [5].
Treatment of Alzheimer's disease
Alzheimer's disease (AD) is a disorder that attacks the central nervous system through progressive degeneration of its neurons. Patients with this disease develop dementia which becomes more severe as the disease progresses. It was suggested that symptoms of AD are caused by decrease of activity of cholinergic neocortical and hippocampal neurons. Treatment of AD by ACh precursors and cholinergic agonists was ineffective or caused severe side effects. ACh hydrolysis by AChE causes termination of cholinergic neurotransmission. Therefore, compounds which inhibit AChE might significantly increase the levels of ACh depleted in AD. Indeed, it was shown that AChE inhibitors improve the cognitive abilities of AD patients at early stages of the disease development. The first generation of AD drugs were AChE inhibitors: alcaloids like (-)-Huperzine A (HupA) and (-)-galanthamine (GAL, Reminyl); synthetic compounds tacrine (Cognex) and rivastigmine (Exelon).
(-)-Huperzine A
(-)-Huperzine A, discovered by Chinese scientists from 1980s, has been proved to be a powerful, highly specific, and reversible inhibitor of AChE. It is a novel alkaloid originally isolated from the Traditional Chinese medicine [1] Qian Ceng Ta which is produced from the whole plant of the firmossHuperzia serrata. Qian Ceng Ta has been used for over 1000 years in China for treatment of contusions, strains, swellings, schizophrenia and myasthenia gravis. Shuangyiping[2], a tablet form of HupA produced from the extracts of Huperzia serrata, was developed in 1996 as a new drug for symptomatic treatment of Alzheimer’s disease in China. Compared with the other three FDA-approved drugs for the treatment of Alzheimer’s disease, Donepezil (Aricept), Rivastigmine (Exelon), Galanthamine (Reminyl), HupA has better penetration through the blood-brain barrier, higher oral bioavailability, and longer duration of AChE inhibitory action. The structure of HupA shows some similarity to other known AChE inhibitors. The molecule is fairly rigid and contains an aromatic system as well as a primary amino group that is probably protonated at physiological pH. Various suggestions have been made with respect to its orientation within the active site of AChE, and with respect to the amino acid residue with which its putative pharmacophoric groups might interact. Solution of the 3D structure of a complex of HupA with AChE would permit unequivocal resolution of this issue and it would also provide a rational basis for structure-related drug design aimed at developing synthetic analogues of HupA with improved therapeutic properties.
The crystal structure of the complex of TcAChE with HupA at 2.5 Å resolution (1vot) was determined in 1997 and it shows an unexpected orientation for the inhibitor with surprisingly few strong direct interactions with protein residues to explain its high affinity. HupA binds to TcAChE at the active site, and its in comparison to ACh. The principal interactions of are including: a direct (colored orange) through a water molecule as a linker at the bottom of the gorge; cation-π interactions between the amino group of (colored green) with the distance between the nitrogen and the centroid of the aromatic rings of 4.8 and 4.7 Å, respectively; at the top of the gorge, hydrogen bonds through two water molecules as linkers formed between the amino group of (colored magenta). An unusually short (~3.0 Å) C-H→O HB has been seen between the ethylidene methyl group of (colored crimson) [3].
Tacrine
[3]. In the X-ray crystal structure of TcAChE/ complex which was determined at 2.8 Å resolution, the tacrine is seen (magenta) bound in the active site of TcAChE (1acj) [6]. ACh (gray) is shown for comparison. In the crystal structure of Torpedo californica acetylcholinesterase (TcAChE) complexed with tacrine (THA), THA's acridine ring is stacked between the aromatic rings of , near the catalytic triad of which consists of S200, E327, H440. When comparing 3 recent complexes of TcAChE, i.e. edrophonium (EDR), decamethonium (DECA) and THA, the only major conformational difference between them is seen in the orientation of the phenyl ring of F330. In the DECA complex it lies parallel to the surface of the gorge; in the other two complexes it is positioned to make contact with the bound ligand. This close interaction was confirmed by photoaffinity labeling by a 3H-labeled photosensitive probe, which labeled, predominantly, F330 within the active site. Labeling of was also observed. One mole of label is incorporated per mole of AChE inactivated, indicating that labeling of W279 and that of F330 are mutually exclusive. The structural and chemical data, together, show the important role of aromatic groups as binding sites for quaternary ligands, and they provide complementary evidence assigning W84 and F330 to the "anionic" subsite of the active site and W279 to the "peripheral" anionic site.
Huprine X
(HUPerzine A + tacRINE) is one of the most potent reversible AChE inhibitors. This consists of a carbobicyclic moiety resembling that of (−)- (colored blueviolet) and the 4-aminoquinoline substructure of (colored magenta). Both these compounds are known AChE inhibitors. (−)-Huperzine A and tacrine positions partially overlap each other at the TcAChE . TcAChE residues interacting with (−)-huperzine A (1vot) are colored orange and with tacrine (1acj) are colored cyan. The of the 4-aminoquinoline substructure of the huprine X in its complex with TcAChE (1e66, TcAChE interacting residues are in green) is very similar to that of tacrine. The ring system of (−)-huperzine A is almost 180° relative to that of huprine X [7].
Galanthamine
[4]. (red) is an alkaloid from the flower snowdrop (Galanthus nivalis). The X-ray crystal structure of the TcAChE/GAL complex (1dx6) was determined at 2.3 Å resolution. The inhibitor binds at the base of the active site gorge of TcAChE, interacting with both the choline-binding site (Trp84) and the acyl-binding pocket (Phe288, Phe290). The tertiary amine appears to make a non-conventional hydrogen bond, via its N-methyl group, to Asp72. The hydroxyl group of the inhibitor makes a strong hydrogen bond (2.7 Å) with Glu199 [8]. ACh (gray) is shown for comparison.
Edrophonium
[5] is stacked between the aromatic rings of , near the TcAChE which consists of S200, E327, and H440 (2ack or 1ax9) [9].


![[2]](http://www.54md.com/drugstore/pic/gpic_25fd25197010a0fb4a680516735e613c.jpg){kind=link}