Computer-Aided Protein Visualization Lab
Knowing the three-dimensional structure of a protein can be a very powerful tool for biologists. Much can be learned about enzyme function, interaction of molecules in your immune system, the appearance of the surface of viruses, and the interaction of ligands and receptors.
One particularly key area of current research is the design of drugs against specific protein targets. Scientists may look for drugs to block the activity of an enzyme of an attacking bacteria, protist or virus (for the purposes of this lab, we are going to focus on SARS-CoV-2). After first finding an enzyme that is at least slightly different between humans and the invading species, the scientists can then use computers to look at the enzyme and try to fit tens of thousands of compounds into the enzyme in such a way as to block its activity; most commonly this involves plugging the active site of the invading species enzyme selectively. Using computers to analyze this problem can speed up the screening of 50,000 potential drugs from many years down to one week or less! Once a few compounds with potential are approved by the computer, the scientist can look to chemically modify those compounds to make them even better and then try them out in drug trials against the enzyme in test tubes,see if they inhibit the ability of the organism to reproduce in cell culture (for the SARS-CoV-2 virus, in particular) and eventually in drug trials in animals and humans.
First some background: (make sure that you understand the bold words)
Proteins are synthesized on ribosomes by linking together many amino acids into a long chain. If you could observe a protein as it is made, it would look like a string of pearls (amino acids) feeding out the end of the ribosome as it floats in the cytoplasm of the cell (Video of Translation (DNALC)). This structure is called the primary structure (or 1° structure) and refers to the sequence of amino acids of the protein. After protein synthesis has started, the sequence of amino acids will begin to fold into a 3-dimensional structure. This process is called protein folding:
Protein folding:
The most important rule about protein structure is that it is determined by the primary sequence of the protein. Protein folding is a complicated multi-step process. The first step results in the secondary structure (or 2o structure) of the protein. Secondary structures come in two flavors: alpha helices and beta sheets (or beta-pleated sheets). Alpha helices are spiral staircase structures (see structure 1 below), and beta-pleated sheets are flat regions where the amino acids run back and forth next to each other in long ribbons (see structure 2 below). These two structures form spontaneously based on the shape/hydrophobicity/charges of the amino acids and are held together by hydrogen bonds. The protein will now look like a string of pearls with twists or zig-zags at intervals along its length.
1. In the structure window, click on "Popup" button to open a larger popup window of this structure. You can toggle the spin of the structure on or off by clicking on the "Spin" button. Clicking and holding on the structure in the window will allow you to manipulate the structure, rotating in three-dimensions.
This is an alpha helix. The amino acid backbone (the parts of the amino acids that are linked together by a peptide bond to form the 1o sequence) is shown in pink/red. The amino acid side chains are shown in yellow (each type of amino acid has its own unique side chain, one of 20 different types). If a section of a protein's primary sequence of amino acids forms this coiled structure, it is known as an alpha-helix.
[Put in manipulations of cartoon view to stick, backbone only then backbone with side chains.......adjust color scheme to emphasize secondary structure. Isolate secondary structures if possible, etc]
2.
The second step of protein folding results in the tertiary structure (or 3° structure). Tertiary structure gives the protein an overall three-dimensional structure. The tertiary structure of a protein is determined by a combination of factors including hydrogen bonds, ionic bonds (between positively and negatively charged amino acids), covalent disulfide bonds (between cysteine residues), and Van der Waals interactions. Tertiary structure can also be affected by repulsive forces between similarly charged amino acids, as well as hydrophobic and hydrophilic interactions with a solvent (commonly water). At a distance many proteins form what look to be large globs at this point, and it is only upon more careful and close up inspection that one can see the true uniqueness of the shape.
 Figure 3: The various intramolecular interactions that help determine tertiary structure
Figure 3: The various intramolecular interactions that help determine tertiary structure
Proteins may contain only alpha helices, only beta sheets, or a combination of the two. The same holds true for the bonds giving a protein its tertiary structure - all, some or none may be present. These different folding patterns existing in different proteins are what give the proteins their distinctive shapes and sizes. A protein that is 300 amino acids long will be 100 nm as an extended chain. If the protein is an alpha helix, it will be 45 nm long; a beta sheet will be 7 x 7 x 0.8 nm; and a small globular form will form a sphere only 4.5 nm in diameter!
Finally, multiple proteins might interact to form a quaternary structure (4°), sometimes known as a protein complex. Each protein in a quaternary structure is called a subunit, and in some cases multiple subunits of the same protein interact to form a quaternary structure (as in the SARS-CoV-2 spike protein you will learn more about below). On the other hands, some quaternary structures contain multiple types of proteins that interact. Most quaternary structures are held together by weak intermolecular bonds (ionic, H-bonds, hydrophobic, van der waals, etc) and occasionally by strong covalent bonds (such as disulfide bonds). While the designation "subunit" is usually reserved for a individual protein that makes up a part of a larger quaternary structure, in some cases term subunit is can also be used to designate large portions of the a protein primary structure that have distinct functions. An example of this alternative use of the term "subunit" is found in the SARS-CoV-2 spike protein (below). Even though each spike protein is made up of three identical subunits (thus is a homotrimer), each of these single polypeptide is further divided into two main subunits--the receptor-binding subunit (S1) and membrane-fusion subunit (S2) (see below).
Domains:
Parts of the secondary and tertiary structures of a protein are usually arranged to form domains, functional units associated with a particular structure. For example, a pair of alpha helices situated side by side might form a binding site, or a particular folding pattern might form the active site of an enzyme, where it binds to its substrate, or the site at which it binds to a coenzyme such as NAD+. The structure of the domain (though not necessarily the exact amino acid sequence) is frequently preserved in different proteins from the same organism that have a similar function (to bind to a cellular receptor, for example, in the SARS-CoV-2 Spike Protein, or to move phosphate groups, for instance). Domains are also conserved in proteins from different species that have the same function (such as hemoglobins for oxygen transport or cytochromes in the electron transfer system of mitochondria). Variations in the amino acid sequences in similar domains (or in the nucleotide sequences or genes that code for the proteins) give important clues about evolutionary relationships between organisms. Individual domains are sometimes found (but not always, a fact that makes this a very controversial topic) contained within single exons of eukaryotic genes (exons and introns are concepts you will learn more about later in the course, when we discuss eukaryotic gene structure). In other words, a single exon might represent all of the protein coding sequence required to generate a functional domain within the context of the whole protein structure. This finding has implications for the evolution of eukaryotic genes, since it implies that new proteins can be generated by simply duplicating preexisting protein domain encoding exons and recombining them into new combinations (a process known as exon-shuffling). Thus, a vast variety of proteins with new functions can be generated from preexisting genes, allowing great evolutionary flexibility. Looking at the genes of many eukaryotic organisms shows that this is exactly what appears to happen.
A good example of all of these principles can be found in the SARS-CoV-2 Spike protein (see figure 4). This is the protein the SARS-CoV-2 virus relies on to gain entrance into its target......that target being any cell that contains the Angiotensin-converting enzyme 2 (ACE2) protein on its surface. The Spike protein is made up of three identical strands of protein arranged into a quaternary structure (a homotrimer). Each is synthesized as an individual protein and then later complexed into the complex secondary, tertiary and quaternary structure you see below. Each individual polypeptide of the spike protein is divided into four major components, S1 (or receptor-binding subunit), S2 (membrane-fusion subunit), TM (trans-membrane anchor) and IC (intracellular-tail). The interaction of secondary structure to form tertiary structure, and the interaction of these structures to form quaternary structure is apparent. The final step of protein folding results in quarternary structure (or 4° structure). This step is only taken in proteins that are made of multiple subunits; meaning that strands of proteins - coded for on separate mRNAs and synthesized independently - come together to form a single functional molecule. Many proteins have multiple subunits; for example, the SAR-CoV-2 protein is made up of three subunits (figure 4).
Figure 4: SARS-CoV-2 Spike Protein representations.
a. 
Schematic drawing of the three-dimensional (3D) structure of SARS-CoV-2 coronavirus spike. S1, receptor-binding subunit; S2, membrane fusion subunit; TM, transmembrane anchor; IC, intracellular tail.
b. 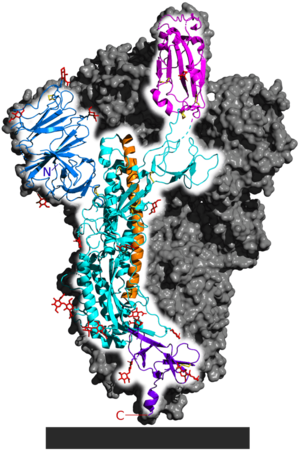
This is the same structure represented in the schematic above, except the 3-dimensional structure of one of the three components of the overall spike protein quaternary structure is shown (the other two members of the homotrimer are grey) . The flat ribbons show beta-sheets while the cylindrical coils represent alpha-helices. The S1 receptor-binding subunit is magenta, while the S2 membrane-fusion subunit consists of the blue, teal, orange and purple portions. The TM and IC domains are not shown.
Determining the 3-Dimensional Structure of a Protein
Scientists can use several techniques to observe the folding of a protein.
(1) The most commonly used technique is called x-ray crystallography. This technique requires the scientist to form crystals of the protein of interest - very similar to how you can form sugar crystals by dangling a string in a super-saturated sucrose solution! The crystal is then bombarded with x-rays, and the diffraction pattern of the x-rays is recorded on film. By analyzing the diffraction pattern, the spacing of atoms in the protein can be determined. Rosalind Franklin also used this technique on DNA crystals; her diffraction pictures were in turn used by James Watson and Francis Crick to determine the double-helix shape of DNA.
(2) A second technique used is NMR, or Nuclear Magnetic Resonance, (also called MRI in medicine). In this technique proteins are placed in a magnetic field. The resonance frequency of the field can be varied. Different atoms in different chemical environments will absorb maximally at different frequencies. By viewing a spectrum of absorbance vs. resonance frequency, it is possible to specify the identity of atoms and their location with the protein. This technique is particularly useful where it can detect movement in molecules as proteins fold and/or as they bind with other molecules.
(3) A third technique becoming more common is Cryogenic electron microscopy (cryo-EM) which, according to Wikipedia, is "an electron microscopy (EM) technique applied on samples cooled to cryogenic temperatures and embedded in an environment of vitreous water. An aqueous sample solution is applied to a grid-mesh and plunge-frozen in liquid ethane or a mixture of liquid ethane and propane" (https://en.wikipedia.org/wiki/Cryogenic_electron_microscopy)
(4) Yet another technique that has been developed is the use of computers to simulate protein folding strategies. Originally programs were developed to allow scientists to predict the structural effect of a relatively small change in a protein sequence. The computer will look at the three-dimensional structure, as determined by x-ray crystallography or NMR, of a closely related protein (a homologue from another species or a slight variant from the same species) and predict what the effect of the amino acid changes would be. This process is done by having the computer determine the "lowest energy configuration" of the protein - or simply put, which folding of the protein puts the least stress on the molecule. It looks to make sure that two amino acids will not be pushing into each other, that two similarly charged amino acids will not be opposing each other, that hydrogen bonds and disulfide bonds are formed where they can be, etc.
New programs aim to predict the three-dimensional structure of proteins from scratch - where no known homologue has ever been studied. This technique is quite powerful because forming crystals of many proteins is hard, if not impossible. Instead, these programs start at the same point that protein folding starts in the cell. They take the primary sequence of the protein and look for the correct sequences of amino acids to form alpha helices and beta-pleated sheets. Once these are in place, the program searches through for tertiary structures that obey the "lowest energy configuration" rules.
Viewing the 3D structure of a protein
This now brings us back to where we started at the top of page 1 - once we know the structure, how can we look at it? There are many computer programs in existence to visualize proteins in three-dimensions. For the drug design studies, powerful computers and programs are necessary to analyze the energetics of drug fit. (Simulating structures and their interactions is a powerful “weeding out” tool in deciding which drugs to test in laboratory studies, making the development of new drugs more efficient and less costly.)
For this lab you will be working with a very commonly used computer modeling program known as Jsmol to look at "pdb" files of proteins with a known structure (using the methods outlined above). Jsmol is an open-source JavaScript viewer for chemical structures in 3D: http://wiki.jmol.org/index.php/JSmol. We are using the website proteopedia.org as a wiki host site to house and access these exercises.
Preliminary Questions
Answer the next few questions to review your organic molecule knowledge and to get up to speed quickly.
1. What are the four major organic molecule groups?
2. How many of the other three groups can you find in association with proteins? Where in a cell do these interactions occur?
3. Draw the structure of an amino acid. Identify the position of the alpha carbon (called ca in the MAGE program). Identify the location of the side chain.
4. What is an enzyme?
5. Draw a schematic figure to show the progression of a protein from simple primary structure to the most complex tertiary structure.
Make sure you answer all questions in the lab. Questions marked by an * are more complicated and may require outside reading and thinking time to answer completely.
Using Jmol through proteopedia.org
A VERY BRIEF GUIDE TO USING Proteopedia
All of the structures hosted these sites initially are viewable in the small window on the right side of the page. However, if you click on "popup" in the lower left of that window, a larger window containing the structure will become visible. This window can be resized to any size by dragging the window edges to desirable dimensions.
TURNING THE PROTEIN: Once you have the molecule displayed on the black screen, try rotating the molecule by placing the pointer on the screen, holding down the mouse button and dragging the mouse. Move the mouse slowly so that you can see the molecule turn. If you place the pointer in the center of the field and drag left/right, the protein will rotate around the y-axis. If you place the pointer in the center of the field and drag up/down, the protein will rotate around the x-axis. If you place the pointer at the top of the field and drag left/right, the protein will rotate around the z-axis.
TO VIEW SELECTED GROUPS (carbon backbones or side chains of amino acids): Make sure the protein is not spinning (click on "toggle spin" in the bottom left corner of the image window to stop spinning). Then, simply move your cursor to any portion of the molecule you want to identify. After a second or two, a small window should popup containing a chain of information--a three letter code (amino acid) followed by a number (the position of the amino acid in the protein primary structure), a chain designation (indicating which protein chain/subunit you are pointing at) and then the element you are pointing at (C,H,N,O,P,S,Zn,Mg,Ca,etc).
Here are the structures you will want to examine
1.Protein 1: MHC Class I
2.Protein 3: HIV Reverse Transcriptase
3.Protein 2: Alkaline Phosphatase



