Introduction
is an important enzyme for the breakdown of triglycerides in the body.
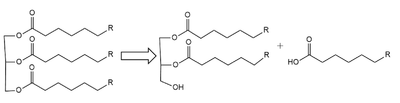
Purpose of LPL: catalyze the breakdown of a triglyceride into a diglyceride and create one free fatty acid
A
lipase is an enzyme that is capable of catalyzing the
hydrolysis of fats/lipids which are consumed through oils. It is encoded by the
p22 region in chromosome 8. Once synthesized, it is secreted into the interstitial space in several tissues. The main site of action for is in the
capillary lumen within muscle and adipose tissues. The function of this lipase is to hydrolyze
triglycerides of very low density lipoproteins (
VLDL) and to aid in the delivery of lipid nutrients to vital tissues. The enzyme is commonly found on the surface of cells that line blood capillaries. Two different lipoproteins are essential to break down triglycerides. One of the lipoproteins is utilized to transport fat into the bloodstream from different organs. The lipoproteins essential, in the transport of fat from the intestine are referred to as
chylomicrons. VLDL are utilized in carrying triglycerides from the liver into the bloodstream. The hydrolysis of triglycerides by lipoprotein lipase results in fat molecules to be used by the body as energy or stored in fatty tissue.
Structural Overview
is assumed to only be active as a , however, previous studies have argued that the lipase can be active in its . (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6442593/) The N-terminal domain of lipoprotein lipase is known to consist of an alpha/beta hydrolase domain, which is composed of six alpha helices and ten beta-strands. This domain creates an . The C-terminal domain of lipoprotein lipase is composed of twelve beta strands which form a .
Mechanism
Lipoprotein Lipase functions to catalyze the hydrolysis of one ester bond of triglycerides in order to remove one fatty acid tail and turn the triglyceride into a diglyceride. It does this by utilizing a simple serine hydrolase mechanism, in which it uses a composed of Asp183, His268, and Ser159 to catalyze the hydrolysis. His268 serves as a base catalyst by deprotonation of Ser159, which can then serve as the nucleophile. The transition state of the catalysis is stabilized by the backbone of Trp82 and Leu160 residues, forming the . The hydrolysis results in the formation of one free fatty acid and a glycerol with two fatty acid tails.
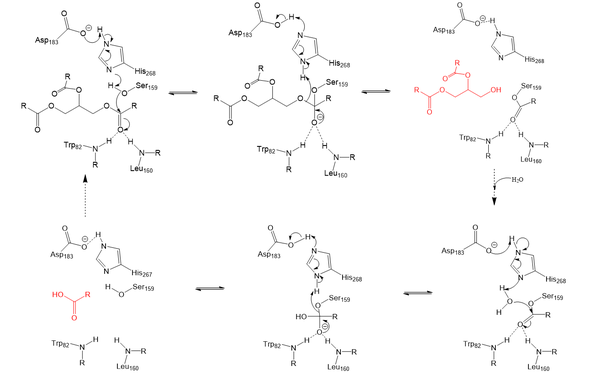
Serine hydrolase mechanism utilized by LPL to catalyze the breakdown of one ester bond of a triglyceride. Compounds colored red are the products of the hydrolysis.
Relevance & Disease
LPL is an extremely important enzyme, in that it is responsible for the proper breakdown of certain fats in the body. LPL breaks down triglycerides carried in chylomicrons, otherwise known as very low-density lipoproteins (VLDL). Chylomicrons carry digested fats in the form of triglycerides out of the small intestine and into the bloodstream. LPL recognizes the chylomicrons, and hydrolyzes the associated triglycerides.[1] When triglycerides are not broken down properly and are allowed to build up, they can lead to increased plasma triglyceride levels (hypertriglyceridemia) and cholesterol buildup. Hypertriglyceridemia is very unhealthy and is the leading cause of Coronary Artery Disease in America.[2] Cholesterol buildup is caused by excess fats (triglycerides) and is a similarly serious issue with regards to obesity and heart disease in the United States as it can lead to plaque buildup in arteries and veins, which restricts blood flow.[3] Chylomicronemia, which is defined as an excess of chylomicrons in the blood, is the disease characterized by the body being deficient in LPL resulting in persistent hypertriglyceridemia. This disease causes the body to be unable to digest very much ingested fats, and often leads to severe abdominal discomfort and several episodes of acute pancreatitis.[4] In short, without LPL in the body, triglycerides are unable to get broken down, and there is a much higher likelihood of developing coronary & metabolic based diseases.
Structural Highlights
GPIHBP1
Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein (GPIHBP1) is necessary for function and stability.
Glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1 (GPIBP1) is a secondary domain that is critical to the stabilization, function, and movement of LPL. The GPIBP1’s highly acidic and intrinsically disordered N-terminal Domain are essential to the binding of LPL’s C-terminal Domain, which is largely done by . The importance of GPIBP1’s affinity to LPL was analyzed by Birrane et al.[5], and it was found that missense mutations of critical residues resulted in high amounts of impairments . It was also concluded that these impairments caused different profound diseases.
Calcium Ion Stabilization
Ions are widely used in proteins and mechanismistic stabilization in many areas of biochemistry. LPL’s tertiary folding is stabilized by a Calcium (Ca2+) ion. The calcium ion shares electron density with surrounding residues in order to orient the protein in its formal state. The is achieved by the calcium ion’s interactions with the following of LPL’s residues: Ala194, Arg197, Ser199, Asp201, and Asp202.
Lid and Lipid Binding Region
In the presence of the GPIHBP1 inhibitor, the and lipid binding region become visible within the structure. As displayed through a study conducted by Arora et. al, in 2019, the lipid-binding region of LPL actively interacts with the known inhibitor in the dimeric form. [6] This was established to be the only time that the homodimeric form was shown as an active lipase. The lid region residues Ile245, Ile249, V251, Ile252, Leu257, Val260, Leu263, and Val264, is found as an open conformation that is composed of two small alpha helices that reach out and away from the protein. The lid and lipid binding region create hydrophobic patches on the surface of lipoprotein lipase which are essential for by LPL.
The novel inhibitor bound between lipid-binding region of one LPL monomer and the catalytic site of the other LPL monomer in the homodimeric form.
References
- ↑ Kersten S. Physiological regulation of lipoprotein lipase. Biochim Biophys Acta. 2014 Jul;1841(7):919-33. doi: 10.1016/j.bbalip.2014.03.013., Epub 2014 Apr 8. PMID:24721265 doi:http://dx.doi.org/10.1016/j.bbalip.2014.03.013
- ↑ Austin MA, Hokanson JE, Edwards KL. Hypertriglyceridemia as a cardiovascular risk factor. Am J Cardiol. 1998 Feb 26;81(4A):7B-12B. doi: 10.1016/s0002-9149(98)00031-9. PMID:9526807 doi:http://dx.doi.org/10.1016/s0002-9149(98)00031-9
- ↑ Kruth HS. Lipoprotein cholesterol and atherosclerosis. Curr Mol Med. 2001 Dec;1(6):633-53. doi: 10.2174/1566524013363212. PMID:11899253 doi:http://dx.doi.org/10.2174/1566524013363212
- ↑ Francis A, Levy Y. [Chylomicronemia syndrome]. Harefuah. 2002 Feb;141(2):201-3, 221, 220. PMID:11905095
- ↑ Birrane G, Beigneux AP, Dwyer B, Strack-Logue B, Kristensen KK, Francone OL, Fong LG, Mertens HDT, Pan CQ, Ploug M, Young SG, Meiyappan M. Structure of the lipoprotein lipase-GPIHBP1 complex that mediates plasma triglyceride hydrolysis. Proc Natl Acad Sci U S A. 2018 Dec 17. pii: 1817984116. doi:, 10.1073/pnas.1817984116. PMID:30559189 doi:http://dx.doi.org/10.1073/pnas.1817984116
- ↑ Arora R, Nimonkar AV, Baird D, Wang C, Chiu CH, Horton PA, Hanrahan S, Cubbon R, Weldon S, Tschantz WR, Mueller S, Brunner R, Lehr P, Meier P, Ottl J, Voznesensky A, Pandey P, Smith TM, Stojanovic A, Flyer A, Benson TE, Romanowski MJ, Trauger JW. Structure of lipoprotein lipase in complex with GPIHBP1. Proc Natl Acad Sci U S A. 2019 May 21;116(21):10360-10365. doi:, 10.1073/pnas.1820171116. Epub 2019 May 9. PMID:31072929 doi:http://dx.doi.org/10.1073/pnas.1820171116
[1]
[2]
[3]
[4]
[5] (LPL GENERAL REFERENCE)
[6] (LPL GENERAL REFERENCE BOOK IF NEEDED)
[7]
[8]
[9]
[10]
Student Contributors
Giselle Flores
Dustin Soe
Maggie Stopa



{kind=link}