From Proteopedia
proteopedia linkproteopedia link Introduction
Esomeprazole is a Proton Pump Inhibitor (PPI) that binds to H+/K+-ATPase and inhibits the secretion of gastric acid from the parietal cells into the lumen of the stomach. Esomeprazole’s commercial brand name, Nexium, is used to treat Gastro-esophageal Reflux Disease (GERD), peptic and gastric ulcers, and Zollinger-Ellison syndrome. Ulcers caused by the bacterium Helicobacter pylori can be treated using Esomeprazole in conjunction with proper antibiotics.1 These result in gastric acid accumulation into the lumen of the stomach from the parietal cells.2 Gastric acid is released through the H+/K+-ATPase pump, which is the final step in acid release.2 Esomeprazole is an irreversible inhibitor of the pump.2
H+/K+-ATPase
![] The H+/K+-ATPase pump is located within the cytoplasmic membrane of resting parietal cells When activated, the ATPase is translocated to the canalicular membrane and begins to pump cytoplasmic H+ into the canalicular space, in exchange for extracellular K+ ions. The H+/K+-ATPase pump regulates the final step in the acid secretion pathway.3 Targeting this enzyme using PPIs is the most effective therapeutic control agent of gastric acid secretion.3 The H+/K+-ATPase pump, located within the cytoplasmic membrane of resting parietal cells, gets translocated upon activation to the canalicular membrane and pumps cytoplasmic H+ into the canalicular space, in exchange for extracellular K+ ions. It is a pro-drug that is protonated twice in the acidic environment of the parietal cell to form the active inhibitor, sulfenamide which forms disulfide bonds with Cys 813 and Cys 892 on the α subunit of the H+/K+-ATPase. Esomeprazole inhibits the final step in the secretion of gastric acid](/wiki/images/thumb/e/e8/Cell.jpg/300px-Cell.jpg)
]
The H+/K+-ATPase pump is located within the cytoplasmic membrane of resting parietal cells When activated, the ATPase is translocated to the canalicular membrane and begins to pump cytoplasmic H+ into the canalicular space, in exchange for extracellular K+ ions. The H+/K+-ATPase pump regulates the final step in the acid secretion pathway.3 Targeting this enzyme using PPIs is the most effective therapeutic control agent of gastric acid secretion.3 The H+/K+-ATPase pump, located within the cytoplasmic membrane of resting parietal cells, gets translocated upon activation to the canalicular membrane and pumps cytoplasmic H+ into the canalicular space, in exchange for extracellular K+ ions. It is a pro-drug that is protonated twice in the acidic environment of the parietal cell to form the active inhibitor, sulfenamide which forms disulfide bonds with Cys 813 and Cys 892 on the α subunit of the H+/K+-ATPase. Esomeprazole inhibits the final step in the secretion of gastric acid
The structure of H+/K+-ATPase is an α,β-heterodimeric enzyme, where the catalytic site is present in the α subunit (Figure 2).7 Four transmembrane segments (TM4, TM5, TM6, and TM8) are located in the α subunit and contain the ion binding region of the enzyme.7 Three domains: A (actuator), N (nucleotide), and P (phosphorylation), are located within the α subunit.7 Binding of ions and ATP to these domains will induce movements in the membrane domain that catalyze ion displacement.7 A group of charged, polar, carboxylic acid amino acids are located in the middle of these transmembrane segments.7 A Lys 791 residue present in TM5 is responsible for providing specificity for H+ transport.7 This residue allows the transport of H+ to the lumen.7 The ammonium group of Lys moves into the H3O+ binding site, and displaces the bound hydronium.7 The movement of Lys occurs due to reorientation of TM4 and TM6, as the conformation changes from E1 to E2P and stabilizes H+ bonding of Lys791 (Scheme 1).7 The β subunit contains six or seven N-linked glycosylation sites, which directs the enzyme to the canalicular membrane.7
H+/K+-ATPase is part of the P-type ATPase enzyme family, and transports cations across the membrane.5 The pump goes against the H+ concentration gradient found in the stomach and is powered through ATP hydrolysis.5 The inorganic Pi produced drives a conformational change in the enzyme and allows release of H+ into the highly acidic environment.5 The enzyme catalyzes this reaction by changing conformation states between E1 and E2 (Scheme 1).6 The cytoplasm facing E1 conformational state has a high affinity for H+ ions, while the lumen facing E2 has a high affinity for K+ ions.6 The reaction begins when a hydronium ion binds to the enzyme on the cytoplasmic surface.6 MgATP will phosphorylate the enzyme at an Asp386 residue in a DKTG amino acid sequence to form the E1~Pi H+ intermediate.6 E1 undergoes a conformational change to form E2, where the ion site is exposed and H+ is released at a pH ~ 1.0.7 Extracellular K+ ions then bind to the same exposed region and the enzyme dephosphorylates.7 An occluded form of the enzyme (trapped) is formed once K+ ions bind; the enzyme de-occludes, reforms the E1 complex, and K+ is released.7
Image:Esomeprazole2d.png Esomeprazole 2D Structure Esomeprazole has two important pyridine and benzimidazole moieties linked through a methylenesulfinyl group.
Image:Pymol.jpg Crystalized Structure of H+/K+-ATPase TM helices 5,6,7,8 are each highlighted in a different color; the majority of the A & B domain within the enzyme, non-catalytic portions of the enzyme, and SCH28080 have been omitted for clarity. The figure depicts the proposed binding sites of the crystallized structure of H+/K+-ATPase. Cys813 and Cys892 are highlighted as the two disulfide bond formation sites. Cys 813 is found between TM5 and TM6 domains while Cys892 is between TM7 and TM8 domains.14 Sulfenamide will interact with these two residues and form disulfide bonds.
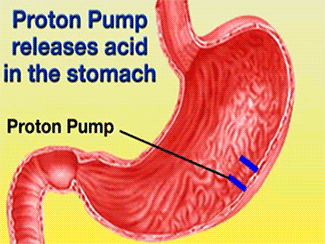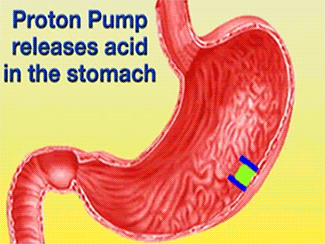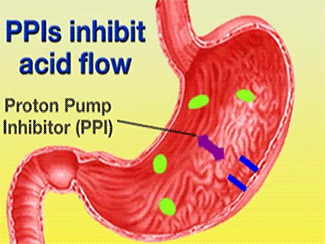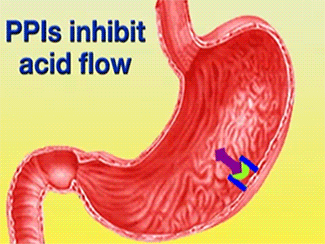
Image:Pymol 2.jpg Binding pocket of SCH28080-ATPase complex PDB image obtained from the RCSB Protein Data Bank.15 Image created using PyMol™ Molecular Graphics System. Majority of the TM domains, non-catalytic portions of the enzyme, and SCH28080 have been omitted. Glu936 (orange), Glu822 (orange), Lys791 (yellow), Glu 795 (orange), Phe126 (gray), Cys892 (light blue), and Cys813 (light blue) are proposed to be part of Esomeprazole’s binding cavity.15,16
Image:Pump.jpg H+/K+-ATPase pump The signals (acetylcholine, histamine, and gastrin) activate the pump in order for the vesicles to move toward the lumen of the stomach.9 These signals bind to their corresponding receptors and activate the cAMP dependent pathway and the Ca2+ dependent pathway.9 Increased levels of intracellular Ca2+ and cAMP will promote the translocation of vesicles to the canalicular membrane, activating the H+/K+-ATPase.9 Histamine binds to a receptor (Histamine H2), and sends a signal through a G protein which activates adenylate cyclase, and catalyzes the conversion of ATP to cAMP.9 Gastrin will stimulate the release of histamine by binding to the gastrin receptor (CCK2).9 Acetylcholine binds to a receptor (Muscarinic M3) and releases Ca2+ from the endoplasmic reticulum.9
Image:Esomeprazole Mechanism.jpg Activation of Esomeprazole to sulfenamide7. R1=OCH3, R2=CH3, R3=CH3, R4=CH3, X=CH, Bz=benzimidazole, Py=pyridine.7 Mechanism: (1) protonation of Py, (2) protonation of Bz, (3) intramolecular rearrangement of BzH+-Py, forms sulfenic acid (4) dehydration to form sulfenamide (5) disulfide bond formation between enzyme Cys residues and sulfenamide.
The mechanism by which Esomeprazole is converted is as follows: the pyridine ring is first protonated (1), which alters the configuration of the enzyme to the E2 form. Esomeprazole accumulates in the stimulated secretory canaliculus of the parietal cell. As H+ is being transported by the ATPase, the second H+ is added onto the benzimidazole moiety (2).7 The bis-protonated forms are in equilibrium with the unprotonated pyridine and protonated benzimidazole rings.7 The protonated benzimidazole ring (electrophilic C2) reacts with the unprotonated pyridine moiety (nucleophilic N) enabling intramolecular rearrangement, resulting in a tetracyclic sulfenic acid (3).7 The sulfenic acid is dehydrated to form an active sulfenamide; both are thiophilic agents that are permanently cationic and membrane impermeable (4).12 Sulfenamide can form disulfide bonds with Cys813 residues located between TM5 and TM6 loops and a Cys892 located between the TM7 and TM8 loops on the α subunit of the H+/K+-ATPase (5).14
The interaction between Esomeprazole and H+/K+-ATPase has not yet been crystallized.15 However, data obtained from the crystallized structure of a SCH28080-ATPase provides structural and binding site information.16 SCH28080 is a competitive K+ inhibitor that interacts with the enzyme’s Phe126 residue and prevents disulfide bond formation between Omeprazole and Cys813.16 This suggests that SCH28080 and Esomeprazole have mutually exclusive inhibitions showing an overlapping binding site.16 The crystallized structure of SCH28080-ATPase shows the same luminal-open (E2) conformation as Esomeprazole, and is the first and only crystallized evidence of PPI-ATPase binding site and conformational change.16 Using this information, the SCH28080-ATPase crystallized structure provides evidence of the two binding sites, Cys813 and Cys892.15,16 The binding pocket, as proposed using SCH28080 includes the following residues: Glu936, Lys791, Glu795, Cys813, Cys892, Phe126, and Glu822 (Figure 5). The negatively charged residues within the TM domains are important for K+ binding and are conserved in all P-type ATPases.16 The SCH28080 model shows that electrostatic and hydrophobic factors affect drug-enzyme interaction.16
![] The H+/K+-ATPase pump is located within the cytoplasmic membrane of resting parietal cells When activated, the ATPase is translocated to the canalicular membrane and begins to pump cytoplasmic H+ into the canalicular space, in exchange for extracellular K+ ions. The H+/K+-ATPase pump regulates the final step in the acid secretion pathway.3 Targeting this enzyme using PPIs is the most effective therapeutic control agent of gastric acid secretion.3 The H+/K+-ATPase pump, located within the cytoplasmic membrane of resting parietal cells, gets translocated upon activation to the canalicular membrane and pumps cytoplasmic H+ into the canalicular space, in exchange for extracellular K+ ions. It is a pro-drug that is protonated twice in the acidic environment of the parietal cell to form the active inhibitor, sulfenamide which forms disulfide bonds with Cys 813 and Cys 892 on the α subunit of the H+/K+-ATPase. Esomeprazole inhibits the final step in the secretion of gastric acid](/wiki/index.php/Image:Cell.jpg)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}