Background
Human G-protein coupled receptor 40 (hGPR40), also known as free fatty acid 1 receptor (FFAR1), is a seven helical transmembrane domain receptor for long-chain free fatty acids that stimulates insulin secretion.[1] Some known fatty acid substrates of hGPR40 include linoleic acid, oleic acid, eicosatrienoic acid, and palmitoleic acid[2]. This enzyme is primarily located in the pancreatic β-cells in the islets of Langerhans, and as such, it has become a target for potential Type II Diabetes treatments.[3] Activation of hGPR40 has been shown to both stimulate insulin secretion and decrease glucose concentration.[4] GPR40 is a member of a group of homologous GPCRs all located on chromosome 19q13.1 including GPR41, 42, and 43.[5]
Structure
Like most G-protein coupled receptors, hGPR40 contains (). To obtain a crystallized structure of the protein, four (, , , ) were made to increase expression levels and thermal stability of the protein. These mutations did not significantly impact the enzyme's binding affinity with a known agonist, TAK-875. A (shown in crimson) was also added to intracellular loop 3 to aid in the formation of crystals. The lysozyme also had little effect on TAK-875 binding.[1] For clarity, the lysozyme is removed in all further renderings of hGPR40.
Binding Sites
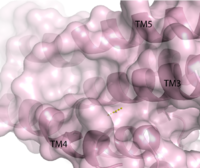
Figure 1. Second proposed binding site of hGPR40.
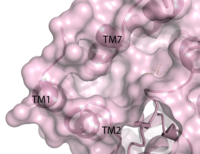
Figure 2. Third proposed binding site of hGPR40.
identified multiple binding sites in hGPR40.
Full agonists and
partial agonists were shown to bind in separate sites with positive
cooperativity.
[6] The has been identified, but other binding sites were hypothesized. TAK-875 binds between transmembrane helices 3, 4, and 5 and underneath ECL2. By visual inspection, a second possible binding site was proposed between transmembrane helices 3, 4, and 5 on the intracellular side of the transmembrane helices. Also by visual inspection, a third possible binding site was proposed between transmembrane helices 1, 2, and 7 on the extracellular side of hGPR40, close to the TAK-875 binding site.
[1]
Charge Network
hGPR40 has a distinct binding pocket that is established by seven key residues. The importance of these residues for agonist binding was determined by
mutagenesis studies. Each of these residues have either a
charged or polar R-group that allows them to develop a charge network. This network keeps the residues in a stable, unbound state until exposed to a substrate. When the substrate (an agonist) enters the binding pocket, four of the seven interact directly with the carboxylate moiety of the agonist. In 2007 and 2009 researchers showed the presence of Arg 183 and Arg 258 in the binding pocket
[7][8] Along with the two Arginine residues, the charge network incorporates two Tyrosine residues.These residues (Tyr 91 and Tyr 240) also stabilize the carboxylate of the agonists. It was further determined that Tyr 240 is epecially important for binding. Mutation of Tyr 240 caused a reduction in the binding affinity of TAK-875 by eight fold and had a significant effect on the
KD of the protein.
[1]
ECL2
Although it may be different in many ways, hGPR40 is similar to most G protein coupled receptors because it contains a highly conserved hairpin loop. This extracellular loop (), is accompanied by a disulfide bond and serves an important role in the protein. In hGPR40, ECL2 has two sections: a beta sheet and an auxiliary loop. The beta sheet (shown in cyan) spans helices 4 and 5. The ECL2 of hGPR40 differs from that of other proteins because it contains an auxiliary loop (magenta) of 13 extra residues. The entire extracellular loop has low mobility and flexibility which allows it to act as a cap for the binding pocket. The only exception to the low flexibility is the tip of the auxiliary loop, which corresponds to residues Asp 152-Asn 155. This area of greater mobility allows for substrates to enter the binding site.[1]
Function
hGPR40 functions as a free fatty acid receptor that participates in insulin signaling to regulate blood glucose concentrations. The actual mechanisms by which this occurs are unknown, but there are multiple theoretical mechanisms for how hGPR40 accomplishes this.
Mechanisms of Insulin Secretion
One proposed pathway of insulin secretion by hGPR40 involves the activation of the Gaq/11 protein complex. This complex then activates phospholipase C (PLC) which in turn phosphorylates phosphatidylinositol 4,5-bisphosphate to inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). IP3 can then mediate the influx of Ca2+ caused by the binding of free fatty acids to hGPR40 by moving into the cytoplasm, binding to the endoplasmic reticulum, and allowing for the release of Ca2+ into the cytosol.[5] This increase in [Ca2+] amplifies the similar increase in [Ca2+] that results from high concentrations of glucose. In this way, hGPR40 mimics glucose dependent insulin secretion.[9]
Another pathway through which hGPR40 may induce insulin expression is through phospholipase D1 (PLD1). When free fatty acids bind to hGPR40, it is able to phosphorylate and therefore activate PLD1. The PLD1 plays a role in controlling the organization of an actin network that plays in role in insulin secretion.[5]
Clinical Relevance
By signaling predominantly through Gaq/11, GPR40 increases intracellular calcium and activates phospholipases to generate diacylglycerols resulting in increased insulin secretion. Synthetic small-molecule agonists of GPR40 enhance insulin secretion in a glucose dependent manner in vitro and in vivo with a mechanism similar to that found with fatty acids. GPR40 agonists have shown efficacy in increasing insulin secretion and lowering blood glucose in rodent models of type 2 diabetes.[5]
TAK-875
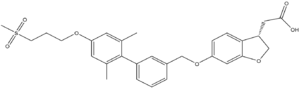
Figure 4. Structure of TAK-875
One example of an hGPR40 agonist is . The carboxylate moiety of the agonist enters through the it of the auxiliary loop, interrupts the charge network, and binds with Arg 183, Arg 258, Tyr 91, and Tyr 240.
[1] TAK-875 has shown efficacy in increasing insulin secretion and lowering blood glucose in rodent models of type 2 diabetes.
[5] This drug was studied in stage III clinical trials and was able to significantly reduce
HbA1c and fasting plasma glucose levels in Japanese patients with type 2 diabetes that was not controlled by diet and exercise. However, clinical trials were stopped shortly after this study because TAK-875 was suspected of causing liver damage.
[10]




{kind=link}