is a multidomain tumour suppressor protein involved in the regulation of various cellular processes, such as cell adhesion, migration or proliferation[1]. It is expressed in plethora of organs and tissues, e. g. cerebral cortex, bronchi or the gastrointestinal tract[2]. Germline truncation mutations of APC result in familial adenomatous polyposis, a hereditary form of colon cancer[3]. Additionally, loss of the C-terminal portion of APC is detected in about 80 % of sporadic colon cancers[4].
The overall structure of APC
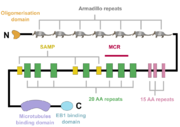
A schematic of the APC protein domain structure. MCR, mutation cluster region; SAMP, Axin-binding motif
The APC protein, its primary sequence encompassing 2843 aminoacids[5], consists of multiple domains, which enable it to interact with diverse partners. At the N-terminus, an oligomerisation domain is found, enabling the APC protein to oligomerise. It is followed by so called pre-ARM region and seven armadillo repeats, which form a groove for binding of a guanine nucleotide exchange factor Asef[6]. The central part of APC contains three 15 aminoacid long repeats followed by seven 20 aminoacid long repeats[1]. These motifs serve as binding sites for β-catenin[7]. In between the 20 aminoacid repeats, three SAMP regions are dispersed, enabling the interaction with Axin[1]. At the C-terminus, a basic domain responsible for binding to microtubules as well as EB1 interaction domain are present[8][1].
Interestingly, majority of somatic mutations occurs in so called mutation cluster region (MCR) between codons 1286 and 1513[9].
The physiological functions of APC and their implications for colorectal cancer onset and progression
Regulation of cell adhesion and migration
The seven armadillo repeats (ARM) together with the so-called pre-ARM region adjoining them at the N-terminus are essential for binding the guanine nucleotide exchange factor Asef[6]. In the absence of APC, Asef adopts an autoinhibited conformation, which prevents it from interaction with the small GTPase Cdc42[10]. Upon APC binding, the autoinhibited conformation of Asef is disrupted and the binding site for Cdc42 is made accessible[6]. Interaction with Asef leads to the exchange of GDP for GTP in the Cdc42 protein, which in turn modulates adherent junctions and contributes to enhanced cell motility[11][10][6]. In colorectal cancers, the truncated version of APC with preserved pre-ARM and ARM domains constitutively activates Asef and hence Cdc42[12]. This leads to extracellular matrix remodelling and promotion of adhesion-independent growth and cell migration[13].
Interestingly, APC takes part in strengthening the adherent junctions through the regulation of cellular distribution of E-cadherin and β-catenin. Full-length APC leads to increased levels of E-cadherin at the plasma membrane and decreases the pool of nuclear β-catenin in favour of the cytosolic one, enabling adherent junctions to be formed[14]. On the other hand, the truncated form of APC lacking the β-catenin interaction motifs is unable of such actions[11].
Regulation of cell proliferation through the Wnt pathway
APC controls cell proliferation as a negative regulator of the Wnt signalling pathway. Together with Axin, GSK3-β, CK1, PP2A and SCFβ-TRCP E3-ubiquitin ligase, APC forms so-called destruction complex, whose role is to promote the degradation of β-catenin[15][16][17]. APC binds Axin through the SAMP regions in APC central part, stabilises it and enables its oligomerisation[18]. Moreover, it has been proposed that APC supports the disociation of phosphorylated (=marked for degradation) β-catenin from the destruction complex[18] and that it is also essential for the movement of the destruction complex towards the plasma membrane[17]. However, degradation of β-catenin is not the only way employed by APC to antagonise the Wnt signalling. β-catenin binding motifs in the central part of APC compete with other interaction partners of β-catenin, such as the transcription factor TCF, thus preventing the expression of pro-proliferative factors (cyclin D, c-myc)[19][20][21][22][23]. Additionally, APC was observed to facilitate the export of β-catenin from the nucleus, hence delivering it out of reach of its target transcription factors[24][25]. Truncated APC defective in β-catenin binding can not effectively control the Wnt pathway signalling either by promoting β-catenin degradation or by preventing it from interacting with transcription factors, which might lead to overactivation of cyclin D or c-myc expression and hence excessive proliferation[19][20][25][18]. However, the first β-catenin binding repeat is often preserved in truncated APC mutants in colorectal carcinomas, which has brought forth the hypothesis that preserving some level of β-catenin downregulation is necessary to balance the potentionally harmful outcomes of constitutive β-catenin activation[26][27][28]

