Introduction
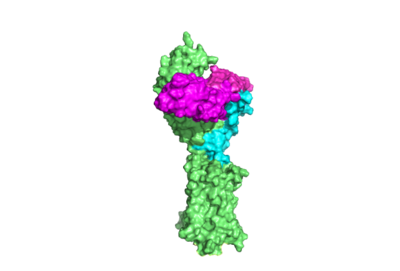
Figure 1. TSHR with TSH bound. The extracellular and transmembrane domains of the GPCR are shown in green, the hinge region in cyan, and thyrotropin bound in magenta.
Thyroid hormones exercise essential functions related to activity of thyroid cells as well as metabolic processes and oxygen consumption[1]. Misregulation of thyroid hormone levels is the cause of many disorders related to hypo- or hyperthyroidism. Thus, understanding the signaling of synthesis and release of these hormones will have applications in treating thyroid hormone disorders[1]. The initiation of the synthesis and release of these hormones is caused by the glycoprotein, thyroid stimulating hormone (TSH), which is released by the anterior pituitary gland[1]. The release of TSH from the anterior pituitary is regulated by thyroid-releasing hormone (TRH) which is released by the hypothalamus. When stimulated by TRH, the anterior pituitary releases TSH. When stimulated by TSH, the thyroid gland will produce and release the the thyroid hormones T4 and T3. T3 is the "active form" of the hormone, however it accounts for only 20% of the thyroid hormone that is released after stimulus by TSH. The T4 that predominates in release from the thyroid will be converted to T3 in the bloodstream. High levels of T3 and T4 can negatively regulate the release of TSH from the anterior pituitary, constituting a negative feedback loop [2]. The thyrotropin receptor is a G-protein coupled receptor on the surface the thyroid gland cells. TSHR is responsible for binding TSH and transduces signal to initiate synthesis and release of thyroid hormones. In addition to TSH, autoantibodies may also bind to TSHR causing inhibition or activation of its desired function. (Figure 1)[3][4]
Structure
Overview
The thyrotropin receptor has an extracellular domain (ECD) that is composed of a as well as a hinge region. This links the ECD to the seven transmembrane helices , which span from the extracellular domain to the intracellular domain [5]. Thyrotropin binding causes a conformational change in the ECD that is transduced through the transmembrane helices. In the active state, the ECD is in the "up" position, while in the inactive state, the ECD is in the "down" state, closer to the cell membrane. A "push-pull" mechanism is proposed to be responsible for the ECD's conformational change between active and inactive states. In the "push" model, TSH binds to the receptor and sterically clashes with the cellular membrane forcing the ECD up away from the membrane. In the pull model, a short α-helix interacts with TSH to pull the ECD up. The active (up) form of the ECD causes a conformation shift in the TMD which causes differential interactions with a heterotrimeric , initiating intracellular signaling[3].
Hinge Region and P10 Peptide
Withing TSHR, the is a scaffold for the attachment of the LRRD to the 7TMD. The hinge region also impacts TSH binding potency and intracellular cyclic adenosine monophosphate (cAMP) levels, mediated by the activation of the GPCR[6]. The hinge region's with the through disulfides. The p10 peptide is a conserved sequence that spans from the last beta sheet of the LRRD to the first transmembrane helix (TM1) and is an intramolecular agonist for conformational shifts in the 7TMD helices[7]. The disulfides between LRRD, hinge helix, and p10 are critical to TSH signaling as they transduce signal from the ECD through the hinge helix to the p10 peptide. Upward movement of the LRRD, caused by TSH binding, will cause rotation of the hinge helix and subsequent movement of the p10 peptide leading to movement of the transmembrane helices which will cause activation of the G-protein. In addition to activation, the hinge region plays an important role in tightly binding TSH. Residues 382-390 of the hinge region adopt a short helix containing two key residues. Y385 from TSHR is buried into a hydrophobic pocket of TSH while D386 forms a salt bridge with R386 of the hormone. that assist in the stable binding of TSH to the TSHR allow more potent activation of the receptor[3]. Even with these key functions, the hinge region itself is not absolutely required for receptor activation[7]. The hinge region functions as a point of attachment to the 7TMD for the LRRD, and its ability to rotate allows for LRRD shifts between up (active state) and down (inactive state) positions. The hinge region also contains key residues that stabilize TSH binding. Disulfides that the hinge helix makes with the LRRD an p10 act as an important communication medium between the extracellular environment and an intramolecular agonist which directly effects conformational shifts in the 7TMD.
7 Transmembrane Helices
The ECD of TSHR is anchored to the membrane through seven transmembrane helices (7TMD), characteristic of GPCRs. Conformational changes in the 7TMD activate intracellular G-protein signaling[5]. Once TSH binds, changes to the p10 peptide are transmitted to the 7TMD. Specifically, hinge helix rotation causes displacement of the p10 peptide that allows the to migrate towards the center of the 7TMD to increase van Der Waals contacts. Additionally, K660 of TM7 forms a stabilizing with E409 of the p10 region. Hinge helix movement also rearranges Y279 relative to I486 on the neighboring , which links two transmembrane helices and is located extracellularly. Substitution of these residues leads to substantial shifts in the activation of the thyrotropin receptor. Structurally guided mutagenic studies have shown that replacing isoleucine with a more sizeable phenylalanine decreases TSH signaling potency[7][8].The sixth transmembrane helix of TSHR moves outward from the center of the 7TMD to facilitate α-helix 5 of the α-subunit of the G protein (Gα)[7][9]. Gα is activated for intracellular signaling when GDP is exchanged for GTP and dissociates from the γ- and β-subunits of the G-protein (Gγ and Gβ) to bind with other target proteins. Activation of the Gα is caused by conformational shifts in the 7TMD and three intracellular loops which directly interact with the G-protein[5]. Conformational shifts in transmembrane helices are the mechanism of changing interactions of the G-protein with the receptor. Activation of the G-protein is caused by these conformational shifts.
Leucine Rich Repeats
The Leucine Rich Repeat Domain (LRRD) is part of the of TSHR and contains . A unique feature of this region is that it is composed entirely of β-pleated sheets. These β-pleated sheets of the LRRD provides a concave binding surface for TSH, including [3]. These interact with in the seatbelt region of TSH forming a salt bridge and initiating the conformational change by pulling on the hinge region of the receptor [10]. This interaction is specific to TSH and TSHR. When other agonists or antagonists bind to the receptor, the change in conformation is a result of different residues interacting, as explained later in the page. The Leucine residues in the LRRD determine ECD folding and which residues are located on the exterior protein and interacting with ligands.
Active and Inactive Form
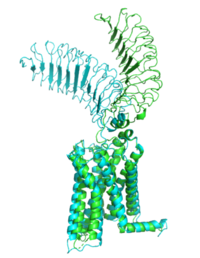
Figure 2: Inactive form of the thyrotropin receptor shown in blue. Active form of the thyrotropin receptor shown in green.
The TSHR protein exists in two states: active and inactive (Figure 2). The protrudes from the cell membrane into the space outside the cell. The contains 7 alpha helices that reside within the cell membrane. The exists when bound to the . One proposed mechanism for the transition from the active to inactive describes that in a natural state, the TSHR ECD can spontaneously transition to the up state, leading to constitutive activity. In this active state, TSH will bind and keep the active state in the up position because of clash with the cell membrane.[10] Conformational change of ECD allows for signal transduction through the TM and into the cell. The ECD rotates 55 degrees up in the active form. [10]
TSHR Agonists and Antagonists
Chemical agonists are found in many living systems and serve as a way to activate receptors or pathways that are necessary for a wide array of biological processes. Chemical antagonists block or inhibit biological processes. Different types of agonists/antagonists exist within the body including hormones, antibodies, and neurotransmitters. The body naturally produces autoantibodies that can act as agonists and mimic the activating mechanism of the natural hormone. Isolating these antibodies in patients with diseases can lead researchers to uncover the mechanism of binding for the receptor.
M22 Agonist and Grave's Disease
is a
monoclonal antibody that produced by patients with
Graves' Disease. Grave's Disease is an autoimmune disease that is a result of hyperthyroidism, where too much TSH is being produced. This disease
effects 1 in 100 Americans and especially women or people older than 30 years of age. The binding of to results in the receptor remaining in its active conformation. In Graves' disease, autoantibodies mimic TSH function and cause thyroid overactivity.
[11]. The M22
autoantibody activates TSHR by causing a membrane clash with the ECD and cell membrane, keeping the TSHR in the active state by preventing the TSHR from rotating to the inactive state (Figure 3). M22 mimics TSH activation of TSHR because it is a potent activator for TSHR.
[10] Although M22 binds in a similar manner to TSH, M22 does not interact with the hinge region when bound to TSHR, whereas TSH bound to TSHR does.
[10] This finding shows that the hinge region is not necessary for the activation of TSHR, and leads to the discovery of other methods of activation.
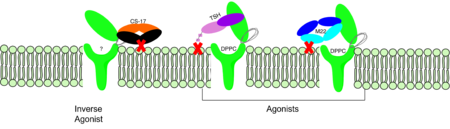
Figure 3: Agonist and antagonist drugs for activating or inactivating the TSHR protein. Here the membrane clashes are demonstrated on TSHR with different agonists attached. CS-17 is orange, TSH is purple, and M22 is blue in the figure. The TSHR protein is green and embedded in the protein.
CS-17 Inverse Agonist
is a monoclonal antibody that acts as an inverse agonist for TSHR constitutive activity. [12]. An example of disease caused by inverse agonists is hypothyroidism. The most common cause of hypothyroidism is Hashimoto’s disease. Without enough TSH to bind TSHR, the pathway remains inactive and thus metabolic processes are inhibited in this pathway. CS-17 interacts with the ECD of the TSHR protein on the convex side GREEN LINK of the LRRD, suppressing TSHR function by keeping the receptor in the inactive state (Figure 3). Clash of bound CS-17 with the cell membrane locks TSHR in the inactive form. This type of inhibition is uncommon and is a promising mechanism for future drug design and research to combat hypothyroidism.[12].
TSH Agonist
This clash is caused by glycosylations of an Asn52 on the . Addition of N-acetyl glucosamine modifications create steric clashes between TSH and the cell membrane, keeping TSHR in the active state.



