UMass Chem 423 Student Projects 2011-1
From Proteopedia
Spring 2011 Chem423 Team Projects: Understanding Drug Mechanisms Instructions posted here: Student Projects for UMass Chemistry 423 Spring 2011 Student projects continued below:
Contents |
Acetylcholinesterase bound by Tacrine
Introduction
Acetylcholinesterase breaks down acetylcholine into acetic acid and choline via a hydrolysis reaction.[1] Acetylcholine is a neurotransmitter that signals muscle contraction. If acetylcholine is not broken down, then the chemical builds up in the synapses between nerve cells and muscle cells resulting in loss of muscle function and ultimately paralysis. The enzyme’s extremely fast reaction rate (approaching the diffusion limit) for breaking one acetylcholine into its two components indicates acetylcholinesterase’s biological importance. Many natural poisons and toxins work by inhibiting this enzyme, thus, paralyzing the victim.[2]
Intentionally inhibiting acetylcholinesterase is a treatment for Alzheimer’s disease. Alzheimer’s is basically the progressive breakdown of the nervous system. Worldwide, there are an estimated 20 million individuals diagnosed with Alzheimer’s, most of whom are over the age of 65.[3] Symptoms of the disease include confusion, irritability and aggressioin, mood swings, language breakdown, long-term memory loss, and eventually loss of bodily functions.[4] To help combat nerve cell degeneration, these acetylcholinesterase inhibitors partially block the enzyme so that excess neurotransmitters remain in the synapse and strengthen the signal.[5]
The drug Tacrine, also known as Cognex, was the first acetylcholinesterase inhibitor approved to treat Alzheimer’s disease. Studies show that the drug only led to slight improvements in people who took it during early stages of the disease, but the drug did nothing to delay the onset of the disease.[6] Tacrine is not often used anymore because it has to be taken four times a day and has adverse side effects, including nausea, diarrhea, heartburn, muscle aches and headaches.[7]
Overall structure
Acetylcholinesterase (AChE) is an monomeric enzyme. Most often, AChE forms a tetramer and binds with a molecule, collagen Q, to connect to the membrane of the neuromuscular junction. [8]. From the , it can be seen that there are 17 and 14 . There are 2 beta sheets formed from 3 anti-parallel and 11 anti-parallel beta sheets, respectively. As the shows, turns, alpha helices, and beta sheets all occupy a portion of the exterior of the protein. The means that the turns must be composed primarily of polar side chains. On the other hand, the alpha helices will be amphipathic with side chain order designated by the helical wheel; the exterior will be filled with polar side chains that can hydrogen bond with water while the inside of the alpha helix will have nonpolar, hydrophobic groups. The beta sheets must also be amphipathic, but the pattern of side chains is alternating polar and nonpolar. In addition, in order to maintain its tertiary structure, the protein has three sulfide bonds, which are covalent bonds that form between cysteine residues. The between cysteine 67 and cysteine 94 is 5.03 angstroms.
Binding
The active site of Torpedo californica acetylcholinesterase (TcAChE) is buried at the bottom of a narrow, deep gorge in the enzyme, and contains a consisting of Ser200, Glu327, and His440. When complexed with tacrine (THA), the aromatic rings of sandwich the THA’s acridine ring . The phenyl ring of Phe330 lies parallel to and in contact with THA. THA is stacked against Trp-84. Its ring nitrogen is H-bonded to the main chain carbonyl oxygen of Hist-440 and it’s amino nitrogen is H-bonded to a water molecule.
Additional Features
Huperzine A reversible binds with AChE at Ser200 but forms hydrogen bonds with Tyr130, Gly117, and Glu199.[9]
Decamethonium is similar to acetylcholine in that it contains trimethylammonium cations allowing it to bind to the nicotinic acetylcholine receptor.[10]
Soman binds to Ser200, in the active gorge. After binding the acetylcholinesterase(AChE) catalyzes the cleavage of the ether bond on the carbon side causing irreversible inhibition.[11]
Rivastigmine reversible inhibits AChE, binding at the active gorge. The AChE cleaves the rivastigmine into carbamyl moiety and NAP.[12]
References
- ↑ http://www.proteopedia.org/wiki/index.php/Acetylcholinesterase
- ↑ http://www.rcsb.org/pdb/101/motm.do?momID=54
- ↑ http://www.searo.who.int/en/Section1174/Section1199/Section1567/Section1823_8066.htm
- ↑ http://en.wikipedia.org/wiki/Alzheimer's_disease
- ↑ http://www.rcsb.org/pdb/101/motm.do?momID=54
- ↑ http://en.wikipedia.org/wiki/Tacrine
- ↑ http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0000930/
- ↑ http://www.ncbi.nlm.nih.gov/pubmed/11804574
- ↑ http://www.proteopedia.org/wiki/index.php/AChE_inhibitors_and_substrates_%28Part_II%29
- ↑ http://en.wikipedia.org/wiki/Decamethonium
- ↑ http://www.proteopedia.org/wiki/index.php/AChE_inhibitors_and_substrates
- ↑ http://www.proteopedia.org/wiki/index.php/AChE_inhibitors_and_substrates_%28Part_III%29
Credits
Introduction - Tyler Vlass
Overall Structure - Zach Brentzel
Drug Binding Site - Andy Kim
Additional Features - Zach Hitzig
Cyclooxygenase
| |||||||
| 3hs5, resolution 2.10Å () | |||||||
|---|---|---|---|---|---|---|---|
| Ligands: | , , , , , , | ||||||
| Gene: | Ptgs2, Cox-2, Cox2, Pghs-b, Tis10 (Mus musculus) | ||||||
| Activity: | Prostaglandin-endoperoxide synthase, with EC number 1.14.99.1 | ||||||
| Related: | 1cvu, 1ddx, 1diy, 5cox, 3hs6, 3hs7 | ||||||
| |||||||
| Resources: | FirstGlance, OCA, RCSB, PDBsum | ||||||
| Coordinates: | save as pdb, mmCIF, xml | ||||||
Introduction
Cyclooxygenase, abbreviated COX, is an enzyme involved in the formation of biological mediators and takes part in the pain and inflammatory response. It serves as an effective pain and inflammation signal in the body to indicate a fault in the body’s homeostatic balance. Drugs target the binding sites of COX to prevent substrate binding and therefore reduce pain and inflammation in the body.
There are two commonly used forms of cyclooxygenase in animals which are denoted as COX-1 and COX-2. COX-1 carries out normal, physiological production of prostaglandins and serves as a basic housekeeping messages throughout the body. Alternatively, COX-2 is constructed in specialty cells and is used in pain and inflammation signaling. COX-2 is “induced by cytokines, mitogens and endotoxins in inflammatory cells, and which is responsible for the production of prostaglandins in inflammation.”
COX has been of research interest because of the value it provides in particular signaling pathways. Currently, cyclooxygenase is widely targeted in the production of a group of drugs called non-steroidal anti-inflammatory drugs (NSAIDS). This group of drugs inclue asprin, ibuprofecn, flurbiprofen and acetaminophen. These drugs attack the binding site of cyclooxygenase and prevent the substrate binding to reduce pain and fever. NSAIDS are broken down into four different classes. Asprin is categorized in class one, ibuprofem is in class two, flurbiprofen and indomethacin are examples of class three, and Vioxx and Celebrex are components of class four. Latest research shows that cyclooxygenase can possibly serve as an effective target in battling cancer including lung and bladder cancer. Additionally, research is being conducted to evaluate the effectiveness of targeting COX in studying Alzhemier’s disease and cardiovascular disease.
Structurally, cyclooxygenase is composed primarily of alpha helices with few beta sheets. The protein contains two binding sites, the cyclooxygenase active site and the peroxidase site.
Overall Structure
COX-2 is a homodimer membrane protein with two identical subunits. The of each subunit contains primarily alpha helices, shown in light blue, with a few beta sheets, shown in yellow. Each subunit contains 587 amino acids.
Each subunit contains three , the epidermal growth factor (red) beginning at the N-terminus, followed by a membrane binding domain (green), and a large catalytic domain at the C-terminus which contains 480 amino acids (blue). The catalytic domain contains two active sites, the cyclooxygenase and peroxidase. The membrane binding domain is made up of four alpha helices. The alpha helices are amphipathic, creating a
, shown in gray, which integrates into the membrane bilayer.
COX-1 and COX-2 are very conserved, being 67% identical in their amino acid sequences. The greatest difference occurs in the membrane binding domain which is only 33% identical.
Drug Binding Site
Many drugs such as aspirin, tylenol, and ibuprofen help regulate pain and and the inflammatory response in the body by blocking the active site of COX. These drugs along with others not only inhibit COX-2 but also inhibit COX-1, causing severe side effects in a small percentage of patients. Recently there has been advances in selectively regulating COX-2 without affecting COX-1 with a drug such as Vioxx. The enzyme COX-2 breaks down arachidonic acid to initiate the production of prostaglandins. Arachidonic acid enters the enzyme through a hydrophobic tunnel formed by the four alpha-helices in the second domain. Once the arachidonic acid reaches the , a hydrogen is believed to be ripped off by the . The heme group located near the peroxidase active site which is believed to help stabilize the radical formed. This radical goes on to further processes that create prostaglandins which intensify pain signals and induce inflammation in damaged parts of the body.
The COX enzyme can be regulated by four different classes of non-steroidal anti-inflammatory drugs (NSAIDS). The first class of inhibitors such as Aspirin regulate the enzyme by irreversibly inactivating the enzyme through covalent modification. A second class of NSAIDS like Motrin or Advil competitively regulates the enzyme. The third class of NSAIDS, for example Flurbiprofen and Indomethacin, forms salt bridges with the enzyme resulting in a slow, time dependent regulation. Finally the fourth class of drugs namely Vioxx and Celebrex selectively regulates COX-2.
Aspirin inhibits the COX enzyme by acetylating the serine residue in the catalytic site preventing the substrate from being catalyzed. Flurbiprofen, a class three inhibitor, binds to the hydrophobic tunnel preventing the substrate from reaching the active site. Flurbiprofen does this with a number of interactions. First it binds to the 120 arginine residue with the formation of a salt bridge. It then hydrogen bonds with the 355 tyrosine residue. An example of a class four inhibitor is SC-558. This drug works in a very similar way as the class three drugs in that it blocks the hydrophobic tunnel but it selectively affects the COX-2. Due to an exposed pocket in COX-2 that is not accessible in COX-1, this drug selectively regulates COX-2 19,000 times more effectively then COX-1. The phenylsulphonamide group of .
Additional Features
Role in other conditions and diseases
COX-2 does not only aid in pain response but plays a role in numerous conditions and diseases including Alzheimer's disease, cardiovascular disease, and cancers such as of the lung and bladder.
Recent discoveries have shown that COX-2 has indirectly played a role in smoker related cancers. Cigarette smoke has been proven to decrease COX-2 expression and cause an increase of PGE2 and TxA2 release. This imbalance causes the progression of tumors and carcinogenesis. This imbalance also contributed to progression of cardiovascular disease. Particularly, more COX-2 positive tumors were found in lung cancer patients than COX-1 positive tumors. Furthermore, smokers of non-cancerous and cancerous patients both had higher expression and imbalance of COX-2, PGE2, and TxA2. Thus, COXIBs are a possible candidate for research in some possible anti-tumor reagents.
Overexpression of COX-2 is observed in later and severe stages of Alzheimer's disease. COX-2 is found to be expressed normally in brain neurons; however, it is questioned as to whether COX-2 prevents or causes neuronal cell death. Some research indicates that an increase in COX-2 and PG synthesis may cause neuronal cell death. Other research has found that NSAID treatment in rats with Alzheimer's disease decreased activated microglial cells and could be used to treat Alzheimer's disease in small doses. Further research must be clarified about the role of COX-2 in the hippocampus, as its role is not clearly defined.
NSAIDS have been shown to have adverse side effects on the kidneys and cause fluid retention,
hypertension, edema, and hyperkalemia through the alterations of renal blood flow, and sodium and potassium
excretion. COX-2 has selective inhibition and is expressed in the kidneys, thus, with NSAID use severe side effects can occur.
References
Wang, Jane; Limburg, David; Graneto, Matthew; Springer, John; Hamper, Joseph; Liao, Subo; Pawlitz, Jennifer; Kurubail, Ravi; Maziasz, Timothy; Talley, John; Kiefer, James; Carter, Jeffrey. The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part 2: The second clinical candidate having a shorter and favorable human half-life. Bioorganic and Medicinal Chemistry Letters. 2010, 20, 7159-7163.
Cyclooxygenase Structure and Mechanism. University of Virginia. http://cti.itc.virginia.edu/~cmg/Demo/pdb/cycox/cycox.html (Accessed April 19, 2011).
Cyclooxygenase. Worldwide Protein Data Bank. http://www.rcsb.org/pdb/static.do?p=education_discussion/molecule_of_the_month/pdb17_1.html (Accessed April 19, 2011).
Kurumbail, R. G.; Stevens, A. M.; Gierse, J. K.; McDonald, J. J.; Stegeman, R. A.; Pak, J. Y.; Gildehaus, D.; Miyashiro, J. M.; Penning, T. D.; Seibert, K.; Isakson, P. C.; Stallings, W. C. Structural basis for selective inhibition of cyclooxygenase-2 by anti-inflammatory agents. Nature. 1996, 384, 644-648.
Luong, C.; Miller, A.; Barnett, J.; Chow, J.; Ramesha, C.; Browner, M. F. Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nat. Struct. Biol. 1996, 3, 927-933.
Messiah College. http://www.messiah.edu/departments/chemistry/molscilab/my_molecules/molslides/my_slides/COX/COX.htm
Chen, George; Huang, Run-Yue. Cigarette smoking, cyclooxygenase-2 pathway and cancer. Biochimica et Biophysica Acta, 2010, 1815, 158-169.
Credits
Introduction: Varun Chalupadi
Overall Structure: Alan Stebbins
Drug Binding Site: Anthony Laviola
Additional Features: Tiffany Brucker
References (edited by): Tiffany Brucker
p38 kinase
| |||||||||
| 1a9u, resolution 2.50Å () | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ligands: | |||||||||
| |||||||||
| |||||||||
| Resources: | FirstGlance, OCA, PDBsum, RCSB | ||||||||
| Coordinates: | save as pdb, mmCIF, xml | ||||||||
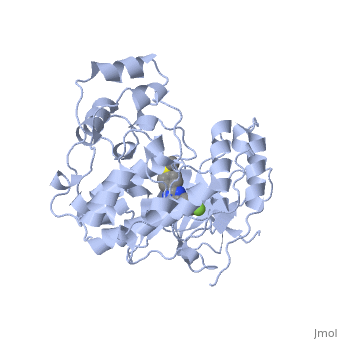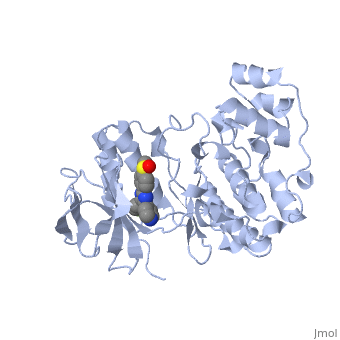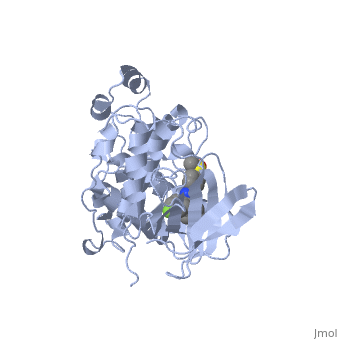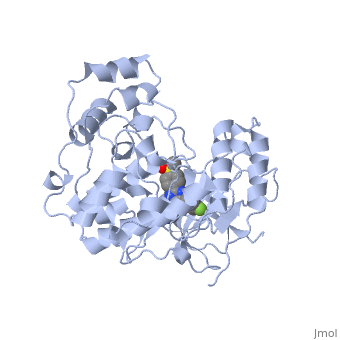

P38 kinase belongs to one of the four subgroups of mitogen-activated protein (MAP) kinases. MAP kinases respond to extracellular stimuli by a signaling cascade leading to intracellular responses. Therefore, MAP kinases function to regulate fundamental cellular processes [1-5].
The p38 subgroup consists of four isoforms: p38α, p38β, p38γ, and p38δ [1-3]. Of these isoforms, p38α and p38β have the most similar amino acid sequences and both forms are expressed in most cell types. P38γ is mostly found in skeletal muscle, while p38δ is only found in the skin, small intestine, pancreas, and kidney [1, 2]. Since p38α was first discovered, most publications focus on this isoform and refer to p38α as p38 [2]. However, all four isoforms have the Thr-Glu-Tyr dual phosphorylation site in the regulatory loop. The substrate specificity of p38 is controlled Glu residue in this dual phosphorylation motif and the length of the loop [1]. This specificity is important for the signal cascade generated in response to stimuli.
Operating as a signal transduction mediator, p38 is activiated by both stress and mitogen stimuli. Environmental stress, particularly UV radiation and osmotic shock, cause an increase activity level of p38. Also, p38 is activated by pro-inflammatory cytokines, especially tumor necrosis factor (TNF) and interleukin-1 (IL1) [3]. However, activation of p38 depends on both the stimulus and the cell type [1]. Dual phosphorylation on the Thr and Tyr is necessary for p38 activation. This dual phosphorylation motif is common in all members of the MAP kinase family. Upstream kinases, which are the MAP kinase kinases (mkks), are responsible for p38 activation [1-3]. Due to selective activation, each p38 isoform requires distinct mkks. Further upstream activators of the MKK/p38 pathway are widely diversified. This cascade accounts for the various stimuli that lead to activating the p38 pathway [1]. Dephosphorylation by dual phosphatases is responsible for the major of the downregulation of p38 [1,3].
The activation of p38 pathway leads to the activation of downstream substrates, such as protein kinases and transcription factors. The p38 pathway regulates close to a hundred genes. P38 is associated with the expression of many cytokines, transcription factors, and cell surface receptors [1]. Various proteins that control transcription and translation are targeted, either directly or indirectly, by p38 kinases. Biological results of p38 activation include inflammation, apoptosis, cell cycle, and cell differentiation [1-3]. However, the role of p38 is specific to cell type [1].
The role of the p38 pathway in cellular inflammation places p38 as a key therapeutic target for inflammatory diseases, cancer, and other diseases. Therefore, p38 inhibitors are key therapeutic agents for the treatment of such diseases. Pyridinyl imidazoles, especially SB203580 (ligand shown in the Jmol diagram), inhibit the catalytic activity of p38 by binding to the ATP site [5]. The ATP binding site provides specificity necessary for highly selective pyridinyl imidazole inhibitors. The inhibitors for p38 do not bind to structurally similar MAP kinases. Other structural factors, such an unique pocket in p38 for the fluorophenyl ring of some pyridinyl imidazole inhibitors, contribute to the selectively of the inhibitor [4]. SB203580 and related inhibitors binds with about equal affinity to both the activated and inactivated forms of p38 kinase. Therefore, binding of the inhibitor can lock p38 into an inactivated conformation.
Gleevec is a brand name drug that targets p38. Gleevec, which is imatinib mesylate, is an inhibitor of p38. Imatinib mesylate, chemically designated as 4-[(4-Methyl-1-piperazinyl)methyl]-N-[4-methyl-3-[[4-(3-pyridinyl)-2-pyrimidinyl]amino]-phenyl]benzamide methanesulfonate [6], is structurally similar to SB203580, 4-[5-(4-fluoro-phenyl)-2-(4-methanesulfinyl-phenyl)-3h-imidazol-4-yl]-pyridine [7]. Gleevec is used to treat certain cancers, including chronic myeloid leukemia, gastrointestinal stromal tumors, and myelodysplastic/myeloproliferative diseases. Gleevec inhibits p38, preventing the proliferation of cancer cells [6].
Overall Structure
Kinase is a single 351 amino acid polypeptide chain made up of 10 alpha helixes and 10 beta strands. Kinase p38 is composed of two domains. The first is a 135 residue N-terminal domain and the second a 225 residue C-terminal domain. The beta strands in light blue form antiparallel beta sheets and are located mainly towards the N-terminus while the alpha helixes in green are located mainly towards the C-terminus. In this rainbow representation of the N and C termini are found at the top of the protein. The catalytic site where the drug binds is located between the two domains.
Drug Binding Site
The p38 kinase-SB2 complex binding chemistry is analyzed in this section. The is connected to the binding site by a hydrogen bond with Met109 and the cyclopropylmethyl group binds to the phosphate-binding ribbon in a depression formed by Val30 and Val38. The complex is also stabilized by contributing from Lys53 and Val105. The conformation of the phosphate-binding ribbon of the p38 kinase changes significantly in order to bind with SB2.
Additional Features
Kinase p38 has a between (also known as domains), which the inhibitors can bind. Blocking p38 kinase may be an valuable way of treating many inflammatory diseases. The pyridinylimidazole inhibitors bind in the ATP binding site, making it competitive against ATP. There has also been observed an allosteric binding site in the Asp-Phe-Gly motif in the active site on p38 kinase for a “diaryl urea class of highly potent and selective inhibitors”. These inhibitors have a new binding mechanism, which does not directly compete with . BIRB 796 is an inhibitor, which has a 12,000-fold increase in binding affinity compared to the previous inhibitor. Structural changes that made such an increase in binding affinity are: methyl substituent on pyrazole ring replaced with tolyl group, chlorophenyl group replaced with naphthyl moiety, and addition of ethoxymorpholine subsituent on paththyl ring. Tolyl has hydrophobic interactions with with side chains of Glu 71 side chain. Glu 71 has one hydrogen bond with the NH on urea. This new conformation is better for supporting the hydrophobic interactions on the tolyl group with the inhibitor. Information about the structure of p38 kinase and its interaction with inhibitors is useful for research about reducing inflammation and diseases such as rheumatoid arthritis. New inhibitors are being designed an optimized to help fight inflammation and other diseases such as rheumatoid arthritis.
Credits
Introduction - Sarena Horava
Overall Structure - Robert Nathan
Drug Binding Site - Nick Cadirov
Additional Features - Inna Brockman
References
1. Ono, K.; J. Han, The p83 signal transduction pathway activation and function, Cellular Signalling 12 (2000) 1-13.
2. Min, L.; B. He; L. Hui, Mitogen-activated protein kinases in hepatocellullar carcinoma development, Seminars in Cancer Biology 21 (2011) 10-20.
3. Raingeaud, J; S. Gupta, et al. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine, The Journal of Biological Chemistry 270 (1995) 7420-7426.
4. Wang, Z.; Canagarajah, et al. Structural basis of inhibitor selectivity in MAP kinases, Structure 6 (1998) 1117-1128.
5. Young, P. R.; M. M. McLaughlin, et al. Pyridinyl imidazole inhibitors of p38 mitogen-activated protein kinase bind in the ATP site, The Journal of Biological Chemistry 272 (1997) 12116-12121.
6. Gleevec http://www.rxlist.com/gleevec-drug-center.htm
7. Ligand Summary for SB2 http://www.pdb.org/pdb/ligand/ligandsummary.do?hetId=SB2&sid=1A9U
http://www.pdb.org/pdb/explore/explore.do?structureId=1A9U
S. Pav, D. M. White, S. Rogers, K. M. Crane, C. L. Cywin, W. Davidson, J. Hopkins, M. L. Brown, C. A. Pargellis & L. Tong. (1997). Crystallization and preliminary crystallographic analysis of recombinant human p38 MAP kinase. Protein Science, 6, 242-245.
L. Tong, S. Pav, D. M. White, S. Rogers, K. M. Crane, C. L. Cywin, M. L. Brown & C. A. Pargellis. (1997). A highly specific inhibitor of human p38 MAP kinase binds in the ATP pocket. Nature Struct. Biol. 4, 311-316.
C. Pargellis, L. Tong, L. Churchill, P.F. Cirillo, T. Gilmore, A.G. Graham, P.M. Grob, E.R. Hickey, N. Moss, S. Pav & J. Regan. (2002). Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nature Struct. Biol. 9, 268-272.
J. Regan, S. Breitfelder, P. Cirillo, T. Gilmore, A.G. Graham, E. Hickey, B. Klaus, J. Madwed, M. Moriak, N. Moss, C. Pargellis, S. Pav, A. Proto, A. Swanimer, L. Tong & C. Torcellini. (2002). Pyrazole urea-based inhibitors of p38 MAP kinase: From lead compound to clinical candidate. J. Med. Chem. 45, 2994-3008.
Drug Binding Site:
Zhulun Wang, Bertram J Canagarajah, Jeffrey C Boehm, Skouki Kassisà, Melanie H Cobb, Peter R Young, Sherin Abdel-Meguid, Jerry L Adams and Elizabeth J Goldsmith. (1998). Structural basis of inhibitor selectivity in MAP kinases. Structure, 6, 1117-1128.
Rituximab Fab
Introduction
| |||||||||
| 2osl, resolution 2.60Å () | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
| |||||||||
| Resources: | FirstGlance, OCA, RCSB, PDBsum | ||||||||
| Coordinates: | save as pdb, mmCIF, xml | ||||||||
Rituximab Fab is used as a prescribed drug to stop transplant rejection, to help regulate autoimmune diseases and to fight cancer. Rituximab has become a key proponent in treatment for cancers such as lymphoma and leukemia and has become the most used chimeric (mouse/human) antibody of its kind. Developed in 1986 by Ivor Royston and Howard Birndorf, Rituximab became FDA approved for clinical use in 1997. Since then Rituximab has become the routine treatment for non-hodgkins lymphoma that otherwise proves to be resistant to certain forms of chemotherapy. Recently, in 2010, Rituximab was approved by the European Commission for use in fighting follicular lymphoma. Because of the wide range of effects that Rituximab causes through its interactions with the CD20 protein, many uses for this antibody have been discovered.
Rituximab is part of a family of antibodies called chimaric monochromal antibodies. These types of antibodies are developed from a single type of parent cell and specifically bind to a single host cell. These types of antibodies do not vary between each other and always interact in the same manner with the chosen cell. Each one of these antibodies is a mixture, chimera, of both rat and human cells both of which play the same role in the host organism. Being chimaric allows for testing on mice before implementation on human cells and combination of human antibodies with mice antibodies makes for simple transitioning between lab testing and clinical testing because the known antibodies have been linked together without making large assumptions with no research.
In brief, Rituximab acts on both benign and malignant lymphocytes, or B-cells. B-cells create antibodies when bombarded with foreign antigens. They are produced in the bone-marrow of humans and when acting normally, help the body fight off disease. If these B-cells are over-produced, under-produced, act abnormally, or are dysfunctional, many different diseases may arise as previously listed. Rituximab acts on the CD20 protein that is found on almost all B-cells. this CD20 protein is unchanged throughout the life of the B-cell.
Multiple proposed mechanisms of action may occur when Rituximab interacts with a lymphocyte (B-cell). The first is known as antibody-dependent cell-mediated cytotoxicity (ADCC) which allows Rituximab to trigger natural cell lysing mechanisms through the use of T-cells and other macrophages. In this proposed mechanism, Rituximab performs as a label to begin cell destruction. Upon further investigation, complement-dependent-cytotoxicity (CDC) became another possible mechanism of action. Through this mechanism, complement proteins are called upon to destroy cell membranes causing overall lysis of the cell. These complement proteins are again naturally occurring, yet usually rely on naturally occurring antibodies to be guided towards non-self antigens. Instead, rituximab acts as the antibody guiding these complements specifically to B-cells. The third and final possible mechanism of action occurs through apoptosis, a more direct route to cell death. Upon initial interaction with the B-cell, the cell triggers programmed cell death. Through this process the cell self-destructs and is immediately destroyed. All three of these mechanisms have been looked into with some uncertainty as to which most likely occurs. Scientists have witnessed many results from treatment such as the shedding of CD23, the down regulation of B-cell receptors, and many other effects that both prove and disprove all three proposed mechanisms.
Rituximab has proven to be a very useful antibody in fighting many human disease, through looking closely at how Rituximab may interact with B-cells, further knowledge about its possible uses will result. This article put together by Chemistry Undergraduate Students at the University of Massachusetts Amherst, will analyze and compile evidence of the chemical mechanisms, binding sites, and applications of Rituximab, a highly used drug in medicine.
Structure
| |||||||||
| 2osl, resolution 2.60Å () | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
| |||||||||
| Resources: | FirstGlance, OCA, RCSB, PDBsum | ||||||||
| Coordinates: | save as pdb, mmCIF, xml | ||||||||
Rituximab is made up of four amino acid chain subunits. Two of these are considered major subunits and two are minor subunits. The two major amino acid sequences are made up of 451 amino acids and the two minor chains contain 213 amino acids. This large protein binds to the surface the surface of B-cells and therefore is exposed primarily to the aqueous external environment. To have stable interactions with the environment, the protein is largely polar, composed almost entirely of beta sheets.
The beta sheets formed in the protein’s native structure form two distinct sides of this protein. Each of these sides is made of woven beta strands that form anti-parallel sheets. The binding site of the protein is found within one of these beta-strand sheets.
Rituximab interacts the surface of its cell membrane through a alpha helix region. This is actually done in two places, where alpha helices of non-polar amino acids are found. Each of these is found at the terminal end of a beta sheet, allowing the active site to bind to the substrate elsewhere on the cell surface.
Each of these structures can be seen in the Jmol native structure found to the right. The red and blue beta strands are the , while the light and dark green strands are the . Each of these has an alpha helix structure, which is shaded in a slightly different color to emphasize its position.
Drug Binding Site
| |||||||||
| 2osl, resolution 2.60Å () | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| |||||||||
| |||||||||
| Resources: | FirstGlance, OCA, RCSB, PDBsum | ||||||||
| Coordinates: | save as pdb, mmCIF, xml | ||||||||
Rituximab (Rituximab Fab) is a chimeric antibody with human IgG1 used in the therapy of non-Hodgkin’s B cell lymphomas. This antibody targets B cells by binding to the cell-surface receptor, CD20. CD20 (human B-lymphocyte-restricted differentiation antigen, Bp35) is a hydrophobic transmembrane protein with a molecular weight of approximately 35 kD located on pre-B and mature B lymphocytes. Rituximab has a binding affinity for the CD20 antigen of approximately 8.0 nM, which is similar to the parent murine antibody, 2B8. The amino acid residues alanine (170) and proline (172) within the extracellular loop of CD20 are critical for rituximab binding. Selection of random libraries yielded 2 distinct peptides binding Rituximab: 1 peptide was homologous to alanine (170)-proline(172), the other was assumed to mimic the same epitope.
Of Rituximab Fab, the Fab (fragment antigen-binding) is the region in which the antibody is bound to the antigen. A heavy and light chain domain located at the terminal end of the monomer shapes the binding site of Rituximab Fab. Each one of these chains (heavy and light) has a constant and variable portion. The constant chain is identical in all antibodies; the variable chain is unique to each specific B-cell antibodies. The heavy chains of Rituximab Fab are made up of approximately 451 amino acids and the light chains by approximately 213 amino acids.
Rituximab can begin complement activation by binding of Clq to the Fc region (region of an antibody that interacts with cell surface receptors) of an antibody when inducing complement-mediated cell lysis. C1q is a 400kDa protein split into 3 subunits. Each subunit consists of Y-shaped triple peptide helices joined at the stem, forming a globular non-helical head at its end. The helical components of the structure contain varied strands of Glycine, proline, isoleucine and/or hydroxylysine. The globular heads of the structure (along with two serine proteases, Clr and Cls) are then responsible for the multivalent attachment of the C1q structure; forming the complex C1 which triggers the compliment cascade that performs cellular lysis.
Additional Effects
Rituximab is used quite frequently to treat dysfunctional leukemias and lymphomas. Rituximab works extremely well to treat these diseases because of the CD20 binding site. This treatment can also lead to an increase in the number of circulating CD20+ B cells. Rituximab has also shown to be effective in the treatment of multiple sclerosis, rheumatoid arthritis, and anemias. However while effectiveness has been proven, there are also concerns about the safety of the treatment. There is current research being conducted in Norway which will research the effectiveness of rituximab to treat chronic fatigue syndrome. Rituximab is also being used to manage the recipients of kidney transplants. All of these treatments are because of the CD20 binding sites.
Credits
Introduction: David Peltier
Structure: Donald Einck
Drug Binding Site: Ethan Leighton
Additional Effects: Chris Coakley
Citations: Chris Coakley
All Green Screen Effects: David Peltier
Citations
- Sieber, S, G Gydnia, W Roth, B Bonavida, and T Efferth. "Combination treatment of malignant B cells using the anti-CD20 antibody rituximab and the anti-malarial artesunate." Int J Oncol. 35.1 (2009): 149-158. Print.
- RITUXAN® (Rituximab) full prescribing information, Genentech, Inc., 2008
- DiJulio JE. Monoclonal antibodies: overview and use in hematologic malignancies. In: Rieger PT, ed.Biotherapy: A Comprehensive Overview. 2nd ed. Sudbury, Mass: Jones and Bartlett Publishers; 2001:283-316.
- Maloney DG, Smith B, Rose A. Rituximab: mechanism of action and resistance. Semin Oncol. 2002; 29(suppl 2):2-9
- Idusogie, Esohe, Leonard Presta, Helene Santoro, Pin Wong, and Michael Mulkerrin. "Mapping of the C1q Binding Site on Rituxan, a Chimeric Antibody with a Human IgG1 Fc." J Immunol. 164.4 (2000): 4178-4184. Print
- Du, Jiamu, Hao Wang, Chen Zhong, Baozhen Peng, and Melian Zhang. "Structural Basis for Recognition of CD20 by Therapeutic Antibody Rituximab." J Biol Chem. 3.27 (2007): 15073-15080. Print
- Edwards J, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close D, Stevens R, Shaw T (2004). "Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis". N Engl J Med 350 (25): 2572–81.doi:10.1056/NEJMoa032534.PMID 15201414
- Braendstrup P, Bjerrum OW, Nielsen OJ, Jensen BA, Clausen NT, Hansen PB, Andersen I, Schmidt K, Andersen TM, Peterslund NA, Birgens HS, Plesner T, Pedersen BB, Hasselbalch HC. Rituximab chimeric anti-CD20 monoclonal antibody treatment for adult refractory idiopathic thrombocytopenic purpura. Am J Hematol 2005;78:275-80
- Patel V, Mihatov N, Cooper N, Stasi R, Cunningham-Rundles S, Bussel JB,Long-term responses seen with rituximab in patients with ITP, Community Oncology Vol. 4 No. 2, February 2007:107
- Polyak MJ, Ayer LM, Szczepek AJ, Deans JP (2003). "A cholesterol-dependent CD20 epitope detected by the FMC7 antibody". Leukemia 17 (7): 1384–9
- Monoclonal antibody FMC7 detects a conformational epitope on the CD20 molecule: evidence from phenotyping after rituxan therapy and transfectant cell analyses. 2001
- Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20 Blood 1994 83:435-445
Drug Binding Site Green Screen Taken from This proteopedia page http://www.proteopedia.org/wiki/index.php/Rituximab
