Post translational modification
BACE1 is synthetised in the endoplasmamic reticulum as an inactive precursor, pro-BACE1. This precursor is then maturated in the Golgi apparatus. It undergoes glycosylation of 4 residues : . This has a role in the activity of BACE1. 3 cysteine residues of the cytosolic tail are also palmitoylated , it is has a role in the localization of the mature enzyme. The propeptide domain is then clived between Arg 45 and Glu 46 of pro-BACE1 by a proprotein convertase (protease). This clivage is known to increase the activity of the enzyme. [2]

Figure 1 : Cleavage of APP by the amyloidogenic and non amyloidogenic pathways
Biological functions
The main biological function of BACE1 is its role in the β amyloid formation by cleavage of the amyloid precursor protein (APP). Indeed the BACE1 cleaves the β secretase site Asp1 or Glu11 of the β amyloid sequence. That initiates the formation of β amyloid. This scission liberates two fragments: a secreted APP ectodomain called APPsβ and a membrane bound carboxy terminal fragment called C99. This C99 will then be cleaved by a ϒ-secretase. This second scission generates the C-terminus of the β amyloid.
In Alzheimer disease, the β amyloid accumulates as fibrillar plaques in the brain of the patient. It triggers the local inflammation, the dysfunction and finally the death of the neurons. BACE1 is so a very important drug target for the inhibition of the β-amyloid production.
It is interesting to note that APP can also be cleaved by α-secretase but, as it does not produce β-amyloid, it is said that it is a non-amyloidogenic pathway (contrary to the amyloidogenic pathway induced by BACE1).
BACE1 may also be involved in other functions in the brain like the regulation of neuronal function, axonal growth, neuroprotection or synapse formation. It could ever have a role in the immunity system as several of its substrates are molecules of the immune system (like IL-1R2). These cross functions need to be investigated before any inhibitor is put to the market.
Structures
Structurally, the 501 amino acid sequence of BACE1 belongs to the eukaryotic aspartic proteases of the pepsin family and contains a bilobal structure constituted of by an N- and a C-terminal domains. BACE is composed of 12 and 28 . Both N- and C-domains are formed by highly twisted β-sheet structures and each domain contributes with an aspartic acid to the catalytic module of the enzyme. The ligands containing a positive charged moiety might then be favorable to counteract the negative charged active site. BACE1 has two aspartic protease active site motifs, DTGS () and DSGT (), and mutation of either aspartic acid renders the enzyme inactive. Like other aspartic proteases, BACE1 has an N-terminal signal sequence (residues 1–21) and a pro-peptide domain (residues 22–45) that are removed post-translationally, so the mature enzyme begins at residue Glu46. Importantly, BACE1 has a single transmembrane domain near its C-terminus (residues 455–480) and a palmitoylated cytoplasmic tail. Thus, BACE1 is a type I membrane protein with a luminal active site, features predicted for β-secretase. The position of the BACE1 active site within the lumen of intracellular compartments provides the correct topological orientation for cleavage of APP at the β-secretase site. As observed with other aspartic proteases, BACE1 has that form three intramolecular disulfide bonds (yellow) and several N-linked glycosylation sites.[1] [3]
In mouse and human brain, native BACE1 is also found as a homodimer. Dimerization is dependent on membrane attachment and increases BACE1 affinity toward APP-like peptides when compared to the monomeric, soluble form. The different enzymatic properties of monomeric and dimeric BACE1 need to be considered in future drug screening and development processes. [4]
Inhibition
Mechanism of inhibition
Structural information about the interaction of substrate with the active site of BACE1 would greatly facilitate the rational design of small molecule BACE1 inhibitors. The molecular modeling identified several residues in BACE1 that potentially contribute to substrate specificity. In particular, forms a saltbridge with the P1' Asp+1 residue of the β-secretase cleavage site, thus explaining the unusual preference of BACE1 among aspartic proteases for substrates that are negatively charged at this position. In addition, several hydrophobic residues in BACE1 form a pocket for the hydrophobic P1 residue. The model also showed that the mutation LysMet→AsnLeu at P2-P1 interacts more favorably with and the hydrophobic pocket of BACE1 than does wild-type substrate, providing an explanation for the enhanced cleavage of this mutation. Conversely, the substitution of Met→Val at P1 blocks the catalytic residue, explaining the lack of cleavage of this mutation by BACE1.
Four hydrogen bonds from the catalytic aspartic acid residues ( and ) and ten additional hydrogen bonds from various residues in the active site are made with the inhibitor, most of which are conserved in other aspartic proteases. The X-ray structure indicates that and the hydrophobic pocket of the active site play an important role in substrate binding, confirming the results of the molecular modelling study. In addition, the bound inhibitor has an unusual kinked conformation from P2' to P4'. The BACE1 X-ray structure suggests that small molecules targeting and the hydrophobic pocket residues should inhibit β-secretase cleavage. Moreover, mimicking the unique P2'-P4' conformation of the bound inhibitor may increase the selectivity of inhibitors for BACE1 over BACE2 and the other aspartic proteases. [1]
Inhibitors
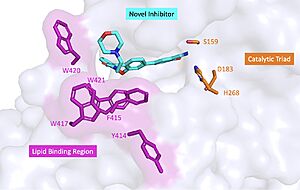
Figure 2 : Docking study on compound 4 bound to BACE1. The coordinates of BACE1 was taken from the crystal structure of 1FKN. The protein is shown in cartoon, while the important residues and ligand 4 are shown in stick model.
In the past, major efforts in designing BACE1 inhibitors were focused on the transition state analogs such as hydroxyethylamines, hydroxyethylene, and tatine-based peptidomimetic inhibitors but their relatively large sizes and excessive number of hydrogen-bond donors and acceptors made it difficult for them to penetrate the blood brain barrier. Therefore the scientific community focused on other coompounds.
One of the discovered compound, showed weak inhibition activity towards BACE1 (about 42% inhibition ratio at the ligand concentration of 100 μM in the fluorescence resonance energy transfer (FRET) assay system). These compound occupied the S1 pocket and the guanidine moiety formed key binding interactions with the two catalytic aspartic acids, and (Figure 2).
Some known BACE1 inhibitors have an an acetylated guanidine group. Some indole acylguanidines were designed by introducing a carbonyl group into the α-position of the guanidine moiety to improve the inhibitor efficiency and the binding with BACE1 was studied.
There is a large hydrophobic sub-site at the top of the guanidine moiety. The acylguanidine formed crucial interactions with two catalytic aspartic acids ( and ) through three hydrogen bonds. The carboxyl oxygen atoms of the acylguanidine formed water-bridged hydrogen bonds with the side chains of and , and a direct hydrogen bond with as well.
A benzyl group extending from the terminus of the guanidine moiety could fill this sub-pocket and thereby potentially increase the binding affinity. The introduction of the simple benzyl group did not enhance the inhibitory activity. However, when a 3,5-dichlorobenyl group was introduced, the potency was improved. The substituted benzyl group occupied the S1 subsite of the substrate binding pocket of BACE1. The carboxyl oxygen atom of acetamide also forms a water-bridged hydrogen bond with the nitrogen atom of the amide group of .
Surprisingly, a compound which has an acetamide group between two chlorine atoms showed a more substantial increase in potency. If two chlorine atoms are remplaced with two hydrogen atoms or reduced the amide to an amine, the resulting compounds displayed much weaker activities against BACE1. The acetamide nitrogen atom forms another hydrogen bond directly with the main chain carboxyl oxygen of . The two chlorine atoms may force the acetamide group adopting a perpendicular angle with respect to the benzyl group, which plays an important role in binding interactions between acetamide and BACE1. Besides, the hydrogen bonds of the inhibitor with residues and induce a semi-closed conformation. Such a conformation of the flap further strengthens the ligand binding to the enzyme.
Analogs were also synthesized based on indole. The indole group pointed toward the back of the S1’ pocket forming a cation-πinteraction with , which appears to contribute further interactions to improve the potency of the inhibitor. [3]


