</scene>'
Introduction
Diacylglycerol acyltransferase () is a membrane protein that synthesizes triacylglycerides from its two substrates diacylglycerol (DAG) and fatty acyl-CoA for dietary fat absorption and fat storage. DGAT synthesizes triacylglycerides from diacylglycerol (DAG) and fatty acyl-CoA. DGAT can be found expressed in the small intestine’s epithelial cells, in the liver where it synthesizes fat for storage, and in the female mammary glands where it produces fat for milk [1]. DGAT is a member of the membrane-bound O- acyltransferases (MBOAT) family [2]. All of the enzymes within this family are transmembrane enzymes that acylate lipids or proteins. MBOAT enzymes have a conserved MBOAT core, a channel-like region that acts as the enzyme’s active site.
Structure
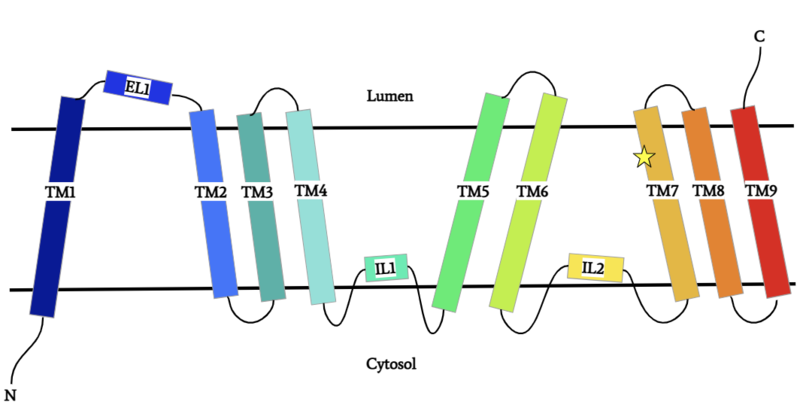
Figure 1: DGAT Structure Overview
DGAT is a dimer that has two identical subunits. Each of the individual subunits contains an MBOAT core that acts as its active site. Each subunit also contains nine transmembrane alpha-helices (TM), 2 intracellular loops (IL), and one ER luminal loop (EL). TM2-9, IL1, and IL2 form the structure of the MBOAT core active site.
Dimer Interface
DGAT is a dimer that has two identical subunits with 9 transmembrane alpha helices (TM), 2 intracellular loops (IL), and one ER luminal loop (EL). The dimer is held together at the dimer interface. Both and hydrophobic interactions between the residues of the TM1 and EL1 regions of both subunits act to hold the subunits of the dimer together.
Acyl-CoA Binding
enters DGAT’s active site through a channel on the cytosolic side of the membrane. In order for the channel to accommodate the fatty acid tail of the Acyl-CoA, His415 must first break its interactions with Met434. This allows for the His415 to swing down and have hydrogen bond interactions with Gln465, thus widening the channel enough for the fatty acid tail to move into the active site. Once , residues Asn378, Gln437, Met434, His415, and Gln465 directly interact with and stabilize the fatty acid tail within the cytosolic channel of the active site.
DAG Binding
enters the active site through the lateral gate located in the lipid bilayer of the membrane. This or lateral gate is a bent and hydrophobic channel that allows for hydrophobic linear or curvilinear molecules to enter. The lateral gate channel is designed to allow for the entrance of DAG and the exit of a triacylglyceride. This channel is also lined with the hydrophobic residues Phe342, Leu261, Val381, and Asn378. Once within the channel, DAG is positioned in close proximity to the bound Acyl-CoA and the catalytic His415.
Active Site
The are Asn378, Gln437, Gln465, His415, and Met434. The active site of DGAT serves its catalytic function by placing the His415 residue in close proximity to the acyl-CoA in order to cleave its ester bond and thus bind the fatty acid to the diacylglycerol. The conserved His415 is able to act catalytically due to a charge relay system, where the neighboring Glu416, due to its negative charge pulls electrons on histidine at the N1 position, making the N3 position more nucleophilic. This nitrogen will then deprotonate DAG so it can begin its attack on Acyl-CoA through acyl substitution. The catalytic mechanism for DGAT is shown in Figure 1.
Disease
Obesity and nonalcoholic fatty liver disease result from an accumulation of triglycerides within the body. Recently, DGAT has become a therapeutic target for obesity and nonalcoholic fatty liver disease in order to reduce triglyceride storage within the body. Different inhibitors have been created, such as AstraZeneca’s direct inhibitor AZD7687 [3]. However, while the triglyceride accumulation decreased, side effects did occur, such as diarrhea and other adverse GI symptoms. Additionally, congenital protein-losing enteropathy (PLE) is a GI disorder that results in the malabsorption of fat and a deficiency in fat-soluble vitamins. There has been a case study of congenital PLE patients that have a homozygous missense Leu295Pro mutation in their DGAT enzymes [4]. This mutation is in a helix of the MBOAT core. Proline is a helix breaker, so it is hypothesized that this mutation breaks a helix in the MBOAT core that impacts its enzymatic activity and ability to make triacylglycerides. Without proper DGAT function to produce triacylglycerides, there is a decrease in albumin, which is a protein that helps prevent fluid leaking out of the liver and blood vessels. This decrease in albumin then leads to decreased efficiency in nutrient transport.
Relevance
[1]
[2]
[5]
[6]
[4]
[3]


