Off-Target Structural Insights: ArnA and AcrB in Bacterial Membrane Protein Cryo-EM Analysis
Mehmet Caliseki, Ufuk Borucu, Sathish K. N. Yadav, Christiane Schaffitzel
and Burak Veli Kabasakal [1]
Molecular Tour
When cells build and maintain their membranes, they need a balance of protein insertion, folding, and degradation. In Escherichia coli, this process is hypothetic to involve the AAA+ protease FtsH, the insertase YidC, and the regulatory HflKC complex. Their interaction had not been shown at structural level. To test this idea, we used single-particle cryo-electron microscopy on detergent-solubilized samples enriched for these proteins.
The results were unexpected. Instead of clear views of an FtsH–HflKC–YidC assembly, the datasets revealed , an enzyme linked to polymyxin resistance, and , the multidrug efflux transporter of the AcrAB–TolC system. Both proteins are known to appear during affinity purification, but their repeated presence across different methods and even in membrane fractions suggests that their recovery was not purely accidental. ArnA, usually described as cytoplasmic, was consistently found in membrane-enriched samples, while AcrB is a well-established membrane protein. We also observed class averages resembling GroEL and cytochrome bo3 oxidase.
These findings show that cryo-EM can capture not only the intended targets but also unexpected complexes that are well resolved and may have physiological importance. While only partial densities of the FtsH AAA+ domain were visible and no stable FtsH–YidC–HflKC complex could be reconstructed, the high-quality ArnA and AcrB structures provide fresh insights into bacterial survival strategies, from antibiotic resistance to drug efflux. More broadly, this study illustrates how structural biology can reveal unplanned discoveries that enrich our understanding of cell biology and the challenges of protein purification.
[[Image:022_Fig2.jpg|thumb|left|250px|Cryo-EM density map and model fitting of the hexameric ArnA complex. (a) Cryo-EM density map of the ArnA hexamer reconstructed at 4.0 A resolution, shown from three orientations: top, side and bottom views. The map reveals the characteristic two-layered architecture of ArnA and clearly resolved secondary-structure elements. (b) Final atomic model of ArnA fitted into the cryo-EM density map. Each of the 12 subunits is displayed in a different color to illustrate the hexameric arrangement. (c) Structural alignment of the final cryo-EM model (blue) with the reference crystal structure(yellow) using ChimeraX. The alignment yielded an rmsd of 1.2 A. Enlarged views show local conformational deviations in loop regions: Glu69–Ala98 in chain D (left) and Pro65–Ser75 in chain F (right). (d) Close-up view showing the model-to-map fit for a peptide segment (Leu483–Arg460). The density mesh is contoured at 1.7 , showing clear peptide backbone density and supporting accurate model placement. 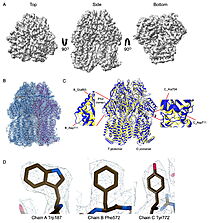 Best fit overlay of the Cɑ positions of SABP2 structures (three structures, light blue carbons) and HbHNL structures (eighteen structures, white carbons) onto the structure of HNL6V (green sticks). The oxyanion hole amide nitrogen atoms of I12 and L81 in HNL6V overlay more closely with the corresponding atoms in HbHNL (I12, C81) than with the corresponding atoms in SABP2 (A13 and L82). |
References
- ↑ doi: https://dx.doi.org/10.1107/S2059798325007089


