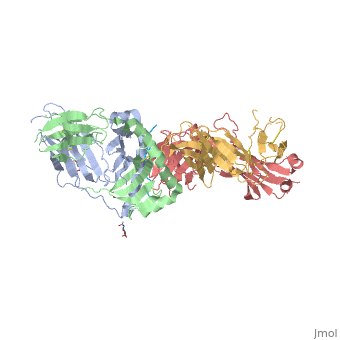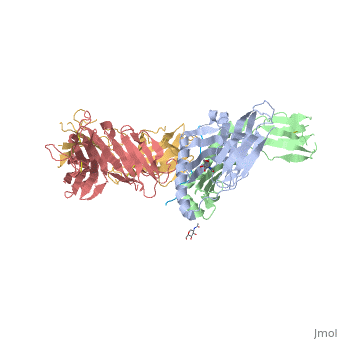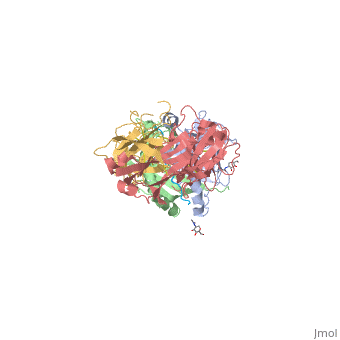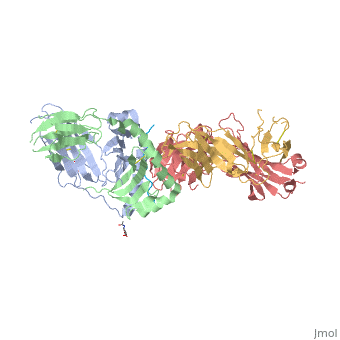
Structure of SP3.4 TCR-DQ8-Glia-αI Complex
The SP3.4 T cell receptor interacts with the DQ8-glia-α1 at 70o across the long axis of the antigen binding cleft. At this antigen binding cleft, the Vα domain of the SP3.4 TCR sits directly above the β helix of HLA-DQ8 and the Vβ domain of the SP3.4 TCR sits directly above the α helix of HLA-DQ8.
The buried surface area (BSA) of SP3.4 TCR-DQ8-glia-α1 interface is 880 Ų. This value BSA displays a standard docking mode to autoimmune TCR-pMHC-II. This indicates this disease is associatd with an autoimmune response and demonstrates how a non self antigen restricted to HLA-DQ8 is identified by SP3.4 TCR.
The CDR loops of the SP3.4 TCR display limited plasticity in the process of docking to the DQ8-glia-α1 complex. On the SP3.4 TCR, the CDR1α loop moves 1.2Å away and CDR3α loop moves 2Å away to engage the DQ8-glia-α1 complex. The slight movement in these particular loops of CDRα may relate to cell signaling. However, the CDRβ loops do not change conformation to promote ligation. Val67 and Arg70 are the only side chains on the DQ8-glia-α1 complex that move to allow ligation with the SP3.4 TCR. Val67 and Arg70 orientate to avoid steric hindrance with Tyr114β and sequentially interact with the CDR3α and CDR3β loops.
The ligation of SP3.4 TCR and DQ8-glia-α1 held together by ten hydrogen bonds and multiple van der Waals interactions. These van der Waals and hydrogen bond interaction between side chains allows for the centrally docking of SP3.4 TCR to HLA-DQ8 during ligation. The interaction between the α chain of the HLA-DQ8 and SP3.4 TCR is entirely due to van der Waals interactions. These van der Waals interactions are stabilized by the CDR3α, CDR1β, and CDR2β loops. The α chain of the HLA-DQ8 is composed of Phe58 to His68. Below the CDR3α loop Phe58 resides and packs against Arg110α due to its aromatic ring. Arg66β is adjacent to the CDR2β loop and interacts with Thr61 and Leu60. Tyr57β of the CDR2β loop packs against Ala64 and points towards His68. His68 interacts with Leu37, which resides in the CDR1β loop.
The ligation of SP3.4 TCR and DQ8-glia-α1 held together by ten hydrogen bonds and multiple van der Waals interactions. These van der Waals and hydrogen bond interaction between side chains allows for the centrally docking of SP3.4 TCR to HLA-DQ8 during ligation. The interaction between the α chain of the HLA-DQ8 and SP3.4 TCR is entirely due to van der Waals interactions. These van der Waals interactions are stabilized by the CDR3α, CDR1β, and CDR2β loops. The α chain of the HLA-DQ8 is composed of Phe58 to His68. Below the CDR3α loop Phe58 resides and packs against Arg110α due to its aromatic ring. Arg66β is adjacent to the CDR2β loop and interacts with Thr61 and Leu60. Tyr57β of the CDR2β loop packs against Ala64 and points towards His68. His68 interacts with Leu37, which resides in the CDR1β loop.
Arg70 resides across the antigen binding cleft and interacts with Gly109α and Thr113β of SP3.4 TCR via van der Waals contact and Hydrogen bonds. The conversion of Leu57α of the CDR2α loop with Thr38α and Tyr38α of the CDR1α loop form a hydrophobic focal point in the β1 helix. This hydrophobic focal point allows for Leu57α Thr38α and Tyr38α to interact with the HLA-DQ8 side chains His81, Ala73, and Thr77.
Interactions with α-I-Gliadin Peptide
The positions P1-Glu, P2-Gly, P3-Ser, P5-Gln, and P8-Gln of the α-I-gliadin peptide interact with the CDR3α, CDR1β, CDR3β and CDR2β loops. Since eight out of the ten Hydrogen bonds present at the SP3.4 TCR-DQ8-glia-αI interface are mediated by the α-I-gliadin peptide residues, the polar interactions with the peptide are the strongest energy contributors to the ligation of SP3.4 TCR to DQ8-glia-αI.
Arg110α of the CDR3α loop has its side chain next to the α-I-gliadin peptide and toward its N terminus. By forming four Hydrogen bonds, the guanidinium functional group interacts with the peptide. The guanidinium functional group also interacts with Phe58α of HLA-DQ8. Before ligation the CDR3α loop is disordered. The stabilization of Arg 110α at ligation causesthe CDR3α loop to become ordered. Arg110α is also stablaized at ligation from an intra chain salt bridge to Asp108α and hydrogen bond with Asp108α to the backbone of Arg110α.
The prominent residues on the gliadin peptide are P5-Gln and P8-Gln. The CDR3β loop is on between these two residues. P5-Gln interacts with the CDR3α and CDR3β loops of SP3.4 TCR. Within this interaction, the P5-Gln forms a hydrogen bond with Arg110α and interacts with Ser111β via van der Waals contacts. P8-Gln interacts with CDR1β, CDR3β and CDR2β loops of SP3.4 TCR. P8-Gln forms hydrogen bonds with the main chain of Val108β of the CDR3β loop and with Tyr57β of the CDR2β loop. P8-Gln also form van der Waals contacts with Ala109β of the CDR3β loop and Leu37β of the CDR1β loop.
Effects of nevirapine binding on the RT-DNA complex
Nevirapine is a first generation NNRTI, which binds RT in a butterfly-like conformation.[1] When DNA binds RT, the polymerase is stabilized by the presence of incoming dNTPs and is said to be in a polymerase-competent state. This is the only way the DNA-RT complex can bind dNTP and incorporate nucleotide. However, nevirapine has a destabilizing effect resulting from decreased interaction of key regions of the polymerase active site with nucleic acid. As previously mentioned, nevirapine leads to opening of the NNRTI binding pocket as a result of switching of the Tyr181 and Tyr188 rotamer conformations and shearing of the β12-β13-β14 sheet away from the β6-β10-β9 sheet.[2] The β6-β10-β9 sheet contains the polymerase "catalytic triad" and the β12-β13-β14 sheet contains the "primer grip" that positions the primer strand for polymerization. Nevirapine binding causes the primer grip to shift by 4 angstroms, lifting the terminus of the DNA primer away from the P site, and diminishing interactions of the primer terminus with the conserved catalytic Y183MDD motif.[2] Therefore, the first effect of nevirapine is loosening of the "primer grip."
The second significant conformational change wrought by nevirapine is on the finger subdomain. The nevirapine-tertiary structure deforms the β3-β4 motif, which usually base pairs with the first template overhang, and interacts with incoming dNTPs that will be incorporated during polymerization. As a result of changes to the β3-β4 motif, parts of the fingers are shifted by 5-7 angstroms into regions that would normally accommodate the template overhang in catalytically-competent RT-DNA molecules.[2] This puts the finger subdomain in an open or semi-open conformation.
Third, the nevirapine-ternary structure extends the conformation of the thumb subdomain making it rigid. This is due to restricted inward movement of the thumb by the displaced β12-β13-β14 sheet.[3]
The crystal structure of nevirapine in solution with RT and DNA showed that the primer grip distortions locked the thumb in a hyper-extended position, resulting in diminished interactions between DNA and the polymerase domain. The flexible fingers, altered template-primer, and shifted template-overhang all reduce the productive binding of dNTPs. It appears that the flexibility of the fingers allow dNTP to still bind the RT-DNA-nevirapine complex at the polymerase active site, however, in a polymerase incompetent mode. The exact mechanism of how dNTPs are able to bind in such a nonproductive manner is yet to be uncovered. The dNTPs may enter the N site and bind in multiple orientations, or they may bind in an ordered yet non-productive fashion once the nucleic acid duplex has slid past the polymerase active site.[4]
HIV-1 RT resistance mutations
Binding of DNA to the polymerase active site requires the primer grip to be near the active site in order for the interaction to be catalytically competent. Binding of nevirapine interferes with the primer grip, moving it away from the active site. A bound nonnucleoside inhibitor is surrounded on three sides: 1) β6-β10-β9 sheet, 2) β12-β13-β14 sheet, and 3) 100-105 loop of p66 + Glu138 loop of p51.[2] Chemical changes in these motifs could lead to the facilitated entrance and exit of most nonnucleosides. In many cases, single amino acid mutations in the binding pocket can significantly decrease the antiviral potency of nevirapine. The mutation K103N confers resistance to a wide variety of nonnucleosides. The Y181C and Y188C mutations also confer drug resistance because they essentially control nonnucleoside movement into and out of the binding pocket depending on the orientations of their side chains. In addition, loop mutations (E138K and K103N) can facilitate exit of nonnucleosides from the binding pocket.[2] To combat this problem, researchers have developed second generation NNRTIs that are generally more effective against a range of drug resistant strains.
Conclusion
Analysis of the crystal structures of RT-DNA complexes in the presence and absence of nevirapine allows us to determine the effects of nonnucleosides on RT structure and function. Binding of nevirapine reveals several changes to the RT-DNA complex. It directly displaces the primer grip located in the palm subdomain, which causes the primer terminus to relocate, the thumb to lock in hyper-extension, decreased interaction between DNA and the polymerase domain, and distortion the dNTP binding site. As a result of the conformational changes nevirapine causes, the process of DNA synthesis by RT is essentially inhibited. Unfortunately, the high mutation rate caused by HIV-1 polymerase's low fidelity readily produces mutant strains that are resistant to NNRTIs. Understanding the precise mechanism of these compounds' inhibition is a key step in developing more effective drugs against HIV.
References
- ↑ Mui PW, Jacober SP, Hargrave KD, Adams J. Crystal structure of nevirapine, a non-nucleoside inhibitor of HIV-1 reverse transcriptase, and computational alignment with a structurally diverse inhibitor. J Med Chem. 1992 Jan;35(1):201-2. PMID:1370694
- ↑ 2.0 2.1 2.2 2.3 2.4 Das K, Martinez SE, Bauman JD, Arnold E. HIV-1 reverse transcriptase complex with DNA and nevirapine reveals non-nucleoside inhibition mechanism. Nat Struct Mol Biol. 2012 Jan 22;19(2):253-9. doi: 10.1038/nsmb.2223. PMID:22266819 doi:10.1038/nsmb.2223
- ↑ Cite error: Invalid
<ref> tag;
no text was provided for refs named kohlstaedt
- ↑ Liu S, Abbondanzieri EA, Rausch JW, Le Grice SF, Zhuang X. Slide into action: dynamic shuttling of HIV reverse transcriptase on nucleic acid substrates. Science. 2008 Nov 14;322(5904):1092-7. PMID:19008444 doi:10.1126/science.1163108




{kind=link}