Introduction
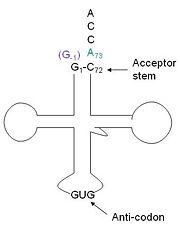
tRNA His, highlighting the incoming G-1 (purple) opposite A73 (green)
tRNAHis has a guanine monophosphate (GMP) residue at the 5’ end in all domains of life, besides α-proteopbacteria. This GMP is referred to as G-1. In prokaryotes G-1 is encoded in the genome. RNase P cleaves pre-tRNAHis to generate the mature tRNA, leaving an extra basepair on the acceptor stem, G-1:C73. In eukaryotes the G-1 residue is not encoded and needs to be added post-transcription. The enzyme that catalyzes this reaction is the polymerase,tRNAHis guanylyltransferase (Thg1). Howerver, the addition of the GMP residue is nontemplated, inserting GMP across from A73 in the acceptor stem creating a mismatch. Unlike most polymerases, Thg1 adds nucleotides in the 3’ –to- 5’ direction, while forming a normal 3’ –to- 5’ phosphodiester bond. Therefore, the 3’-OH of the incoming nucleotide attacks the 5’ end of the polynucleotide chain. This is a two step mechanism where the polynucleotide chain is first adenylated and then guanylated.[1][2][3]
This addition is interesting for multiple reasons. It is one of only a few known reactions where a normal 3’-to-5’ phosphodiester bond is formed in a 3’ –to- 5’ direction. Also, the additional 5’ nucleotide is unique to tRNAHis, with the exception of a tRNAPhe species. Lastly, this modification is essential, at least in yeast.[4] The first crystal structure of Thg1 was the 2.3Å human Thg1 solved in 2010.[5]
Homology
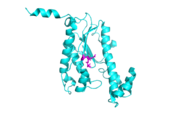
The fold of Thg1 (chain b) is highlighted here with the conserved carboxylates shown as sticks.
3otb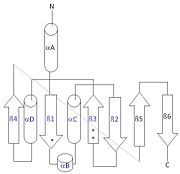
This 2D topology diagram shows the βαββαβ fold of Thg1. The helices and strands involved in the fold are in blue font. The fold is most similar to that of cylcases. The mechanism is more likely the same as family A polymerases, with the conserved carboxylates shown as asterisks(*).
3otbInterestingly, Thg1 shares structural similarities to both cyclases and the palm domain of canonical polymerases, without sequence similarities. The βαββαβ motif is most homologous with adenylyl and guanylyl cyclases. However, based upon the model the mechanism seems to be more like that of a family A polymerase. The model suggests Thg1 has three catalytic : aspartate 29, aspartate 76, and glutamate 77. Cyclases only have two catalytic carboxylates, two aspartates and either cysteine, alanine, or glycine. The position of the carboxylates in Thg1 is homologous to those of T7 DNA Polymerase. An overlay of the palm domain of T7 and Thg1 shows that the three carboxylates, two metal ions, and the incoming nucleotide are conserved and in similar postions. This indicates that Thg1 most likely uses the two-metal-ion mechaism of canonical 5' to 3' polymerases.[6]
Structure
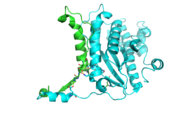
The dimer interface is highlighted between monomers, chain A (green) & chain B (cyan), showing a large contact area. Part of chain A was removed to show more clearly the extensive interface between the monomers. The two salt bridges, between K95 to D128 and E13 to R30, are highlighted as well as some hydrogen bonding.
3otb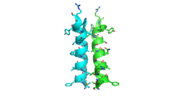
The interface between alpha helices D of the two monomers shows the large amount of contacts helping to stablize the dimer.
3otbEach monomer has an antiparallel β-sheet with six strands and seven α-helices around the sheet. Thg1 forms a homotetramer, with extensive contacts between the . Even though there are fewer contacts between the dimer of dimers, the tetramer is the most stable oligomeric form.
The dimer is stabilized mainly by hydrogen bonds from αD and β4. There are also two salt bridges that help hold the dimer together: Lys to Asp and Glu to Arg (chain A to B). [7]
The reason for the tetramer remained elusive until the "Candida albicans" Thg1 with tRNA His was solved. The model showed that two tRNA His molecules are coordinated between the , each interacting with three monomers. The acceptor stem of the tRNA is coordinated by the dimer of monomers, and the anitcodon is recognized by the second dimer. The binding to the third subunit, is necessary for recognition but may also aid in the correct positioning for guanylylation. Thg1 is selective to binding tRNA His through the recognition of the anticodon loop with base stacking and hydrogen bonding. G34 is recognized via aromatic stacking, between Phe194 and G37(tRNA), this interaction is not base specific but it is purine specific. Asn202 coordinates U35 through hydrogen bonding and His154 base stacks with the third base G36. These interactions cause distortion of the loop that is stabilized by hydrogen bonds from other crucial surrounding amino acids. The structure with tRNA also lends insight into why Thg1 works in is reverse. It appears that Thg1 is a mirror image of forward polymerases, suggesting that the direction in which the template approaches the active site is important in the direction of addition.[8]
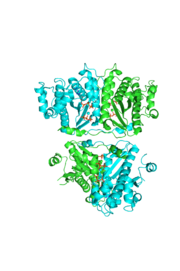
The hThg1 tetramer is shown in cartoon form. The dimer of dimers is crystallographic. The dGTP and triphosphate are shown in sticks, with the coordination metal ions as balls.
3otb Structural highlights
N-terminal helix cap
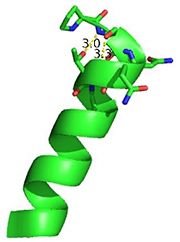
The N-terminal capping motif of αD is examined.
3otbThe N-terminal cap of the helix follows a Ib motif. This motif is also known as a capping box.
N’ -> N4 h-xpxph
N’- P S N Q T L –N4 (residues 135-140 chain A 3otb)
Hydrogen bonds between N’ and the backbone of N3 and N3 with N’ backbone are shown in the figure. The figure is difficult to see the T with P bb but it is not linear, this may just be due to modeling as it is close enough to form a h-bond. There is also a hydrophobic interaction between P and L.
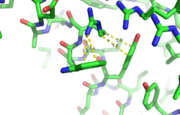
This image depicts two cation-π interactions between Arg and Tyr or Trp. The energetic significances are -1.22 and -6.55 kj/mol respectively.
[9] 3otbA cation-π interaction occurs between a cation and the face of a simple aromatic, there is partial negative charge in the center of the ring. The cation-π interaction is actually stronger than a salt bridge because of the desolvation penalty. With the cation-π interaction the cation has a similar dosolvation penalty to pay as the salt bridge ions but the π system is already poorly solvated. Also there is not neutralization of charge that occurs between the two groups. These properties of the cation-π interaction imply that thecation-π interactions on protein surfaces (mainly where they are seen) could contribute to protein structure and stability.
The Ramachandran plot shows that most of the amino acids follow Ramachadran's restraints. The three that are questionable, N32, D67, S75 are all located in turns.
This is a sample scene created with SAT to by Group, and another to make of the protein. You can make your own scenes on SAT starting from scratch or loading and editing one of these sample scenes.









