metabotropic Glutamate Receptor 5 Homo sapiens
Introduction
G-coupled protein receptors GPCR's are trans-membrane proteins that are integral to cell signaling. The human genome encodes for approximately 750 GPCR's, 350 of which are known to respond to extracellular ligands[1]. GPCR's are divided into four major classes based on sequence similarity and transduction mechanism: Class A,B,C, and F[2]. Metabotropic Glutamate Receptor 5 (mGlu5) is a class C GPCR that is involved in the Gq pathway[3]. mGlu5 is highly expressed in neuronal and glial cells in the central nervous system, where glutamate serves as the major neurotransmitter. When glutamate binds to the extracellular domain of mGlu5 consisting of the Venus Fly Trap motif[4], a conformational change through the trans-membrane domains activates the coupled G-protein. This G-protein disassociates and the alpha subunit activates Phospholipase C which has the final outcome of increased neuronal activity[2].
Structure
Overall Stucture
mGlu5 is seen as a homodimer in vivo, with each subunit being comprised of three domains: extracellular, trans-membrane and cysteine-rich. The structures shown here are centered on the trans-membrane domain, comprised of seven α-helices all roughly parallel to one another[4]. Also displayed is the Intracellular Loop (ICL) 1 which forms a short α-helix. Additionally, ICL3 and Extracellular Loops (ECL) 1 and 3 all lack secondary structure, and ECL2 interacts with trans-membrane (TM) helices 1,2, and 3 as well as ECL 1[4].
Key Interactions
A number of intramolecular interactions within the trans-membrane domain stabilize the inactive conformation of mGlu5. The first of these interactions is an ionic interaction, termed the , between Lysine 665 of TM3 and Glutamate 770 of TM6. Evidence for the importance of this interaction came through a kinetic study of mutant proteins where both residues were separately mutated to alanine, resulting in constitutive activity of the GPCR and its coupled pathway[4]. A second critical interaction that stabilizes the inactive conformer is a between Serine 614 of ICL1 and Arginine 668 of TM3. Similarly, when Serine 614 was mutated to alanine, high levels of activity were seen in the mutant GPCR[4].
A between Cysteine 644 of TM3 and Cysteine 733 of ECL2 is critical at anchoring ECL2 and is highly conserved across Class C GPCR’s[4]. The ECL2 position combined with the helical bundle of the trans-membrane domain creates a that restricts entrance to the allosteric binding site within the seven trans-membrane α-helices. This restricted entrance has no effect on the natural ligand, glutamate, as it binds to the extracellular domain, but this entrance dictates potential drug targets that act through allosteric modulation[4].
Comparison with Class A and B GPCRs
The structure of the trans-membrane domain of mGlu5 was compared to rhodopsin, a class A GPCR, and CRF1R, a class B GPCR. The superimposition of all three structures demonstrated the greatest divergence occurred across the top half of the trans-membrane bundle, attributing to the different extracellular domains that accompany each GPCR class. Furthermore, mGlu5 TM7 is shifted inwards 5 Å compared to the class A GPCR, and TM5 is shifted inwards 6 Å compared to both GPCRs, thus narrowing the allosteric binding entrance[4].
Clinical Relevance
Role in Diseases
mGlu5 is located mainly post-synaptically and is in high abundance in the nucleus accumbens, caudate nucleus, striatum, hippocampus and cerebellar cortex[5].These areas of the brain are highly involved in cognition, motivation, and emotion, which are essential neural functions for everyday life. Diseases and other mental deficiencies arise from either an overactivation of the GPCR, which overactivates its coupled signaling pathway, or from underactivation of both the GPCR and its coupled pathway. Negative allosteric modulators (NAMs) work to decrease protein activity and are being studied as treatments for fragile X-syndrome, depression, anxiety and dyskinesia. Conversely, positive allosteric modulators (PAMs) work to increase protein activity and are being studied for the treatment of schizophrenia and cognitive disorders[6].
Interactions with a Negative Allosteric Modulator
The structure of mGlu5 bound to the NAM Mavoglurant demonstrates how protein activity is decreased through drug interactions.
binds within the allosteric binding site in the core of the seven trans-membrane α-helices, having passed through the restricted entrance formed by the . Bound Mavoglurant forms multiple interactions with the protein that further stabilize the inactive conformation.
The bicyclic ring system of the drug is surrounded by a pocket of mainly hydrophobic residues including Val 806, Met 802, Phe 788, Trp 785, Leu 744, Ile 651, Pro 655, and Asn 747[4] (Figure 1). The carbamate tail of Mavoglurant forms a hydrogen bond through its carbonyl oxygen to the amide side-chain of Asparagine 747 of TM4 (Figure 2). A hydroxyl group similarly forms hydrogen bonds to mGlu5, specifically at two serine residues (S805 and S809) of TM7. These residues form a hydrogen bonding network to other residues through their main chain atoms and a coordinated water molecule (omitted for clarity) (Figure 3). The interactions between Mavoglurant and mGlu5 involve TM helices that were not previously stabilized by any strong interactions, introducing a new level of stability that favors the inactive conformation of the protein and hence decreases the overall activity of mGlu5[4].
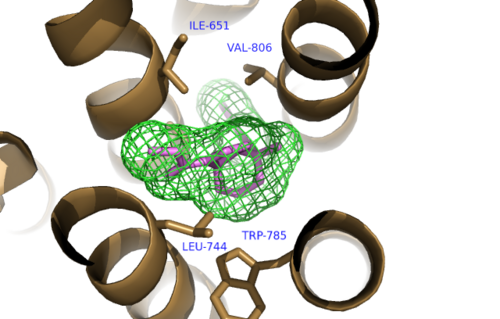
Figure 1. Hydrophobic Pocket Surrounding Mavoglurant
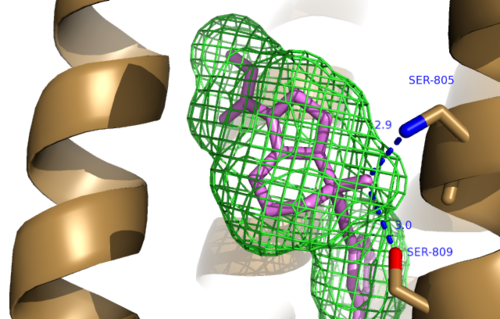
Figure 3. Further Hydrogen Bonding between mGlu
5 and Mavoglurant



{kind=link}