Introduction
Human GPR40 receptor, hGPR40, is a free fatty-acid g-protein coupled receptor that binds medium to long chain free fatty acids, inducing insulin secretion. hGPR40 is highly expressed in human pancreatic β cells, brain, and endocrine cells of the gastrointestinal tract [1]. hGPR40 is of particular interest because the triggering of insulin secrection is glucose dependent.This glucose-dependence for hGPR40 signaling could offer better glycemic control in the treatment of type-2 diabetes.
Function
GPR40 is most prevalent in pancreatic β-cells where free fatty acids (FFAs) have pleiotropic effects [2]. While acute intake of FFAs stimulates insulin release, chronic exposure to high levels of FFAs results in the impairment of β-cell function and insulin secretory response [2]. GPR40 mediates the effect of both acute and chronic levels of FFAs. FFAs amplify glucose-stimulated insulin secretion from pancreatic β-cells by activating GPR40.
When GPR40 is inhibited, insulin secretion no longer increases in response to fatty acid stimulation [2]. This decreased activity of GPR40 leads to a decreased risk of hyperinsulinemia, fatty liver disease, hypertriglyceridemia, hyperglycemia, and glucose tolerance in obese patients [2]. On the contrary, overexpression of GPR40 leads to impaired β-cell function, hyperinsulinemia, and diabetes [2]. These results suggest that GPR40 plays an important role in the mechanism that links obesity and type 2 diabetes and thus is a popular drug target being studied.
Structure
hGPR40 is composed of seven-transmembrane helices that are characteristic of G-protein coupled receptors (GPCR). While there is relatively low sequence identity between hGPR40 and peptide-binding GPCRs and opioid receptors, they do share structural similarities such as a conserved hairpin loop motif on extracellular loop 2 (ECL2) as well as a disulphide bond that is formed between transmembrane helix 3 and the C-terminus of ECL2. Compared to peptide-binding and opioid GPCRs which have distinctive B-sheets spanning from transmembrane helix 4 to 5, hGPR40 possesses a shorter B-sheet-like region which has low B-factors. This reflects the low mobility of the region that limits the overall flexibility of the adjacent portion of ECL2 between Leu171 and Asp175 which forms a cap over the canonical binding site covering it from the central extracellular region.
Ligand Binding
Free Fatty Acids
GPR40’s natural substrate are FFAs in which a free carboxyl group is required to bind. However, GPR40 can be activated by a wide variety of fatty acids with chain lengths ranging from saturated fatty acids with 8 carbons to 23 carbons. In addition, various mono (i.e. palmitoleic (C16:1) and oleic (C18:1) acids) and poly-unsaturated fatty acids (i.e.linoleic (C18:2) and eicosatrienoic (C20:3) acids) can activate GPR40 [3]. The agonists potency varies according to the carbon-chain length however. The activity of GPR40 increases when the chain is increased from C6 to C15 but then decreased when the chain was extended beyond C15. One explanation for this is that as alkyl chain increased, so did the hydrophobic interactions with the protein within the binding pocket. However, for FFAs with carbon chains longer than C15, the molecular size is too large for the binding pocket. This causes the alkyl chain to extend beyond the binding pocket and destabilize the binding [4].
FFAs bind to hGPR40 by coordinating its free carboxyl group to three amino acids, , which are located close to the of hGPR40 on TM5, 6 and 7. Because of the close proximity of these to the extracellular domain and the dominantly hydrophobic nature of FFA’s, it is possible that ligand binding occurs close to, or within, the plane of the membrane [3].
TAK-875
Tak-875 is known to be a partial agonist of GPR40. The binding of TAK-875 to the binding site is fairly unique, as it is proposed that the ligand must enter through the membrane bilayer. This is performed via a method similar to ligand binding to sphingosine 1-phosphate receptor 1 3v2w, retinal loading of opsin 4j4q and the entry of anandamide in cannabinoid receptors, in which block the binding from the extracellular matrix [5]. In contrast, delta opioid receptor binding 4ej4 allow for binding directly from the extracellular matrix. The binding mechanism through the bilayer may be selectively favoring the free fatty acid because of the non-polar regions of the ligand.
TAK-875 binds to a non-canonical site created between transmembrane (TM) domains 3-5 and the extracellular loop 2 (ECL2) of hGPR40. The ECL2 and auxiliary loop form a roof causing TAK-875 to enter through TM3 and TM4, first passing through the lipid bilayer. A disulfide bond is created between Cys79 and Cys170 which increases the rigidity of the ECL2 and allows it to stay in place and function as a cap to the binding site. The carboxylate of TAK-875 is buried within a very hydrophobic region where participates in a complex involving Glu172, Ser187, Asn241, and Asn 244 within the binding pocket of hGPR40 forming ionic and polar interactions with Arg183, Arg258, Tyr91, and Tyr240.
Image:Binding.jpg TAK-875 buries its polar head group within a very hydrophobic region and coordinate within the polar charge network of hGPR40.
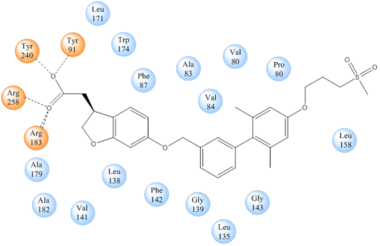
Stabilizing binding interactions between TAK-875 and hGPR40. Amino acids denoted in orange coordinate with the polar head of TAK-875 by polar/ionic interactions while blue amino acids stabilize the binding through hydrophobic interactions.
Signal Transduction
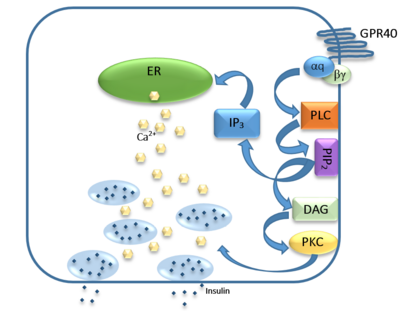
Figure 1: Signaling mechanism mediated by GPR40 in pancreatic β-cells.
The natural substrate of hGPR40 is free fatty acids (FFAs) which bind to the G-protein and enhance glucose-stimulated insulin secretion [3]. FFAs bind to GPR40 which then couples with the G-protein Gq leading to increased phospholipase C (PLC) activity[3]. PLC catalyzes the hydrolysis of the phospholipid phosphatidylinositol-4,5-biphosphate (PIP2) resulting in the formation of diacylglycerol (DAG) and inositol 1,4,5-triphosphate (IP3)[3]. DAG can then activate protein kinase C (PKC) to enhance insulin secretion[3]. IP3 on the other hand is soluble and diffuses to the endoplasmic reticulum where it binds a ligand-gated Ca2+ channel[3]. This binding triggers the opening of the channel causing stored Ca2+ to be released into the cytoplasm[3]. This large increase in intracellular free Ca2+ produces a proportional increase in glucose-dependent insulin secretion, suggesting that insulin release can be contributed in part to the changes in Ca2+ concentration resulting from activated GPR40 [3].
Medical Relevance
While undergoing clinical trials, the use of TAK-875 in the treatment of diabetes mellitus was terminated in the III phase. This was due to observed liver toxicity. The liver toxicity is believed to be due to its effects on the bile acids, achieved through the inhibition of the efflux of bile acids into bile [6]. Other molecules are currently undergoing development in an effort to activate hGPR40 in a way that does not adversely affect the liver. The leading research suggests a molecule similar to that of 3-ethoxypropanoic acid [7]. However, this molecule must be modified before it could be conceivably used, as its half-life is extremely short, due to the rapid oxidation at the benzyl position during metabolism [7]



{kind=link}
{kind=link}