Gamma Secretase
Introduction
Background
Gamma Secretase (GS) is a transmembrane aspartatic protease. It catalyzes peptide bond hydrolysis of type I integral membrane proteins such as Notch, amyloid precursor protein (APP), and various other substrates. It recognizes and catalyzes the reaction with its substrate using 3 residue segments. These 3 residue segments give rise to specific cleavage points for each substrate. APP is the primary substrate of focus, and it is cleaved to form amyloid-β peptides (Aβ). This product is important for various neural processes, and it is well known for its implications with Alzheimer's disease (AD). This has made GS a popular drug target, specifically using gamma secretase inhibitors. However, due to the nature of the enzyme having various neural functions, there are dangerous side effects when it is inhibited.
Overall Structure
GS is composed of 20 transmembrane components (TMs) and has 4 separate protein subunits: Nicastran (NCT), Presenilin (PS1), Anterior Pharynx-defective 1 (APH-1), and Presenilin Enhancer 2 (PEN-2). These subunits are stabilized as functional GS by hydrophobic interactions and 4 phosphatidylcholines.These have interfaces between: PS1 and PEN-2, APH-1 and PS1, APH-1 and NCT.
has a large extracellular domain and one TM. NCT is important to substrate recognition and binding.
serves as the active site of the protease and contains 9 TMs, each varying in length. The site of autocatalytic cleavage is located between of PS1 and a major conformational changes take place in this subunit upon substrate binding.
serves as a scaffold for anchoring and supporting the flexible conformational changes of PS1.
Activation of the active site is dependent on the binding of . PEN-2 is also important in maturation of the enzyme.[1]
Structural highlights
Substrate Structure
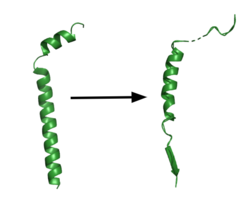
Figure 1. APP fragment conformational change in gamma secretase. APP bound to GS undergoes a conformational change. The free state consists of 2 helices. Once bound to GS, the N-terminal helix unfolds into a coil and the C-terminal helix unwinds into a β-strand. This β-strand interacts with PS1 and is the site of cleavage by the protease.
GS has been structurally characterized in the presence of both APP and Notch substrates. In each of these structures, the substrate bound in a similar location and underwent a similar structural transition upon binding to the active site of GS. Each substrate is composed of an N-terminal loop and a transmembrane helix. The peptide substrate enters the enzyme by via the lid complex, and once in place, the TM helix of the substrate is anchored by . Upon binding to GS, the C-terminal extracellular helix of the substrate unwinds. The substrate's N-terminal end of the TM helix unwinds into a β-strand (Fig. 1). To differentiate substrates, the β-strand is often the main point of identification for the enzyme. Substrate binding induces a structural change in GS, creating two β-strands that form a β-sheet with the one β-strand of the substrate. This β-sheet is in close proximity with the active site, and guides the process of catalysis.[2]
Lid Complex
The is the first point of entry and recognition for the substrate. This lid is located within the NCT subunit between Asn55 and Asn435. This lobe of NCT is divided into two separate subunits, being the large and small lobes. Phe287 from the large lobe acts as pivot between them. This of the small subunit, and these residues compose a greasy pocket that provides an environment for easy movement. The lid consists of 5 aromatic residues which are highly involved with stabilizing the closed conformation. In particular, this conformation is stabilized by . Once the substrate binds and the lid is opened, a charged, hydrophilic pocket is revealed. This pocket contains , and is further involved with substrate binding and recognition once the lid is removed. However, this lid complex is relatively far away from the catalytic site of the enzyme in PS1. Once the substrate binds, the enzyme undergoes a conformational change to shorten this distance, and the rotation of the large lobe in relation to the small lobe reorients the substrate for cleavage by aligning the pocket in NCT to the active site in PS1. [3]
Active Site
The is located between TM6 and TM7 of the PS1 subunit, which is mainly hydrophilic and disordered. Both TM6 and TM7 contribute an aspartate residue to the active site. These two aspartates, are located approximately 10.6 A˚ apart when inactive.[3] Substrate recognition is controlled by the closely spaced PAL sequence of . GS becomes active upon substrate binding, when TM2 and TM6 each rotate about 15 degrees to more closely associate. Two β-strands are induced in PS1, creating an with the β-strand of the substrate.[2] The β-strand of the substrate interacts via main chain H-bonds with the PAL sequence, stabilizing the active site. Asp257 and Asp385 hydrogen bond to each other and are located 6–7 Å away from the scissile peptide bond of the substrate, allowing catalysis to occur.[1] GS cleaves in 3 residue segments which is driven by the presence of three amino acid binding pockets in the active site. [4]
In APP, the cleavage site is between the helix and the N-terminal β-strand. GS can cleave via different pathways, depending on its starting point, but the 2 most commonly used pathways produce Aβ48 and Aβ49.[4]. Tripeptide cleavage starting between results in Aβ48. Cleavage between yields Aβ49. The accumulation of these Aβ peptides has strong implications in Alzheimer's disease.[2]
Relevance
Gamma secretase has been determined to be highly involved with diseases such as Alzheimer's disease (AD). In this, beta-amyloid build up leads to amyloidplaques in brain.[5] These plaques then go on to cause severe neural dysfunction over time. Inhibition of GS could be potential AD treatment, but as stated earlier, this is a hard model to accomplish as the protease is relevant with several different substrates. Complete inhibition would cause other severe implications beyond that of AD, making treatment more difficult than what meets the eye. However, what is known is that there are many different regions that give rise to GS malfunction when they are mutated. Over 200 of these mutations have been linked to causing AD. In particular, these mutations target so called "hot spots" on the enzyme and heavily impact the interface between PS1 and APP, affecting the integrity of catalysis and ultimately creating the plaques that impair neural function. Currently, in order to combat this complex situation, differences in binding between different substrates are being utilized to create drugs that selectively inhibit APP binding with GS, and possibly create a more ideal target for AD treatment[2].

