Titin, one of the largest human protein, in its longest isoform, has a molecular weight exceeding 3 MDa and is over 1.5 μm in length. Titin typically contains immunoglobulin (Ig) domains which are typically 110 amino acids in length, contain an internal disulfide bond and two layers of β-pleated sheets.[1] On the cellular level, titin is typically located within the nucleus of the cell; however, it can also be located within the cytoplasm. [2]
Function
Titin is a highly complex protein responsible for a variety of functions including the key component in the assembly and operation of vertebrate striated muscles. Titin provides connections at the level of individual micro-filaments and contributes to the fine balance of forces between the two halves of the sarcomere.
Titin plays a vital role in the highly ordered macromolecular complex of the sarcomere structures and functions requiring the controlled integration of striated myofibrils in differentiating myocytes. The giant titin protein extends over on half of the sarcomeric unit.
In non-muscle cells, titin plays a role in chromosome condensation and chromosome segregation during mitosis.
Protein kinase, including titin kinase, is a vital aspect of controlling cell proliferation and cell differentiation. Titin kinase is expressed in muscles and is responsible for the interaction with thick filaments known as myosin filaments. The enzymatic activity of protein kinases must be highly regulated through the phosphorylation of specific residues located in the activation component of the catalytic domain. Titin kinase is regulated in a two-step process including the partial unfolding of an inhibitory segment to expose the catalytic region followed by the phosphorylation of the Tyrosin residue. This tyrosine residue is depicted in the structural highlights listed below. [3],[4]
Disease
Myopathy, myofibrillar, 9, with early respiratory failure:
This disease is characterized by adult onset of weakness in proximal (shoulder, upper arm, pelvic area and thighs), distal (lower arms/legs, hands and feet), axial (trunk and head) and respiratory muscles. The main symptoms of onset are pelvic girdle and neck weakness. Ultimately, the weakness will affect the proximal compartment of both the upper and lower limbs. Additional symptoms include varying degrees of Achilles tendon contractures, spinal rigidity and muscle hypertrophy. In extreme cases, respiratory involvement will often lead to the requirement for non-invasive treatment. The natural variant indicating this disease can be found at position 279 and it disrupts NBR1-binding. [5]
Cardiomyopathy, familial hypertrophic 9:
This disease is a hereditary heart disorder characterized by ventricular hypertrophy. The hypertrophy is usually asymmetrical and often involves the interventricular septum (lower heart chambers). The symptoms of this disease include: difficult/labored breathing, fainting, collapse, palpitations and chest pains. These symptoms are often directly agitated by exercise or exertion. The disorder has inter- and intrafamilial variability ranging from benign to malignant forms. Ultimately, this disease has a high risk of cardiac failure and sudden cardiac death. This disease is characterized by a variant in position 740. [6]
Cardiomyopathy, dilated 1G:
This disorder is characterized by ventricular dilation and impaired systolic function. This disorder results in congestive heart failure and arrhythmia, ultimately leading to a high possibility of premature death. This disease is often indicated by natural variants in the following locations: 54, 743, 976, 3799, 4465 or 32996. All of these variants affect the interaction with the TCAP/telethonin. [7]
Tardive tibial muscular dystrophy:
This disease is a late-onset, autosomal dominant distal myopathy. Symptoms are typically muscle weakness and atrophy that are typically confined to the anterior compartment of the lower leg. Clinical onset of this disease usually occur at 35 to 45 years or later. The natural variant of this disease occurs at positions 34306 and 34315. [8]
Muscular dystrophy, limb-girdle, autosomal recessive 10:
This disease is characterized by progressive weakness of the pelvic and shoulder girdle muscles. Muscular dystrophy is an autosomal recessive denerative myopathy that results in severe disability observed within 20 years of its onset. [8]
Salih myopathy:
This myyopathy is an autosomal recessive, early-onset muscular disorder. This disease is characterized by dilated cardiomyopathy, delayed motor development with generalized muscle weakness predominantly affecting proximal and distal lower limbs. Minicore-like lesions with mitochondrial depletion and sarcomere disorganization and cenralized nuclei are present in the skeletal muscle biopsies of affected individuals. Cardiac muscle biopsies display a disruption of myocardial architecture, nuclear hypertrophy, and endomysial fibrosis. This disease can result in sudden death. Mutagenesis occurs in positions 32207 and 32341 and disrupts catalytic activity. [9]
Relevance
Titin is a flexible filament containing a beaded substructure indicating the presence of multiple domains within the molecule. These multiple domains include: the immunoglobulin domain, the Fibronectin type-II domain, the PEVK (proline-glutamate-valine-lysine-enriched unique sequence region, the unique sequences and the kinase domain. [10]
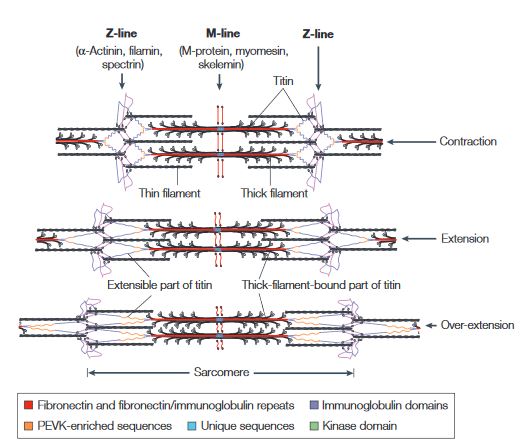
This figure illustrates the ability of titin to coil and extend during muscle contraction and extension.[10]
When the muscle contracts, the sarcomere length decreases resulting in the coiling of the I-band part of the titin. As the muscle extends, the sarcomere length increases resulting in the extension of titin. In the event of over-extension of the muscles there is unravelling of the titin peptide, starting in the least mechanically stable PEVK domain. [10]
Structural highlights
•This is the version of the titin molecule. This structure is colored to differentiate each chain, starting with the blue 5' amino end, ending with the red 3' carboxyl end.
•This secondary structure of titin highlights the of the titin molecule. In this representation, Polar sections of titin are shaded in purple and hydrophobic regions are shaded in grey. The central beta-sandwich structure of the molecule encloses a well defined hydrophobic core. This helps to stabilize the molecule that contains no disulfide bridges and rely solely on hydrogen bonding in the side chains and backbone. Trp34 is also highlighted in this representation to display the central position of the elongated hydrophobic core formed between the two β sheets of the classical Ig folded domain. [11]
•This alternate structure highlights the involved in activity regulation. Full activation of the protein kinase domain requires both phosphorylation of Tyrosine to prevent it from blocking the catalytic aspartate residue, and binding of the C-terminal regulatory tail of the molecule which results in ATP binding to the kinase. [4]
•This structure view highlights the .The VAL residue located at #54 is one of the mutations present in the cardiomyopathy,familial hypertrophic 9, disease. This VAL residue is replaced by a MET residue when the disease is present in an infected individual. [6]
•This is the structure. This secondary view shows multiple titin proteins connected together. This representation is known as the titin band.
•These are the located in the PEVK domain that is vital to the elasticity characteristics of titin.[10]

