This old version of Proteopedia is provided for student assignments while the new version is undergoing repairs. Content and edits done in this old version of Proteopedia after March 1, 2026 will eventually be lost when it is retired in about June of 2026.
Apply for new accounts at the new Proteopedia. Your logins will work in both the old and new versions.
1m10
From Proteopedia
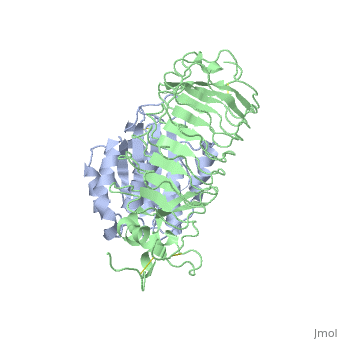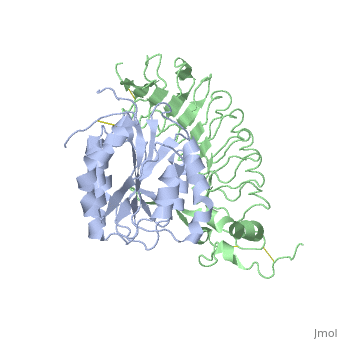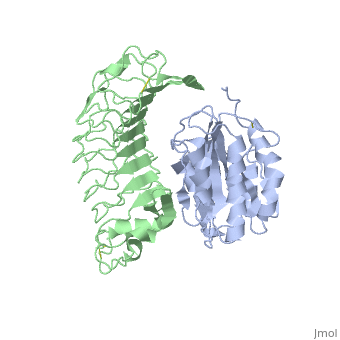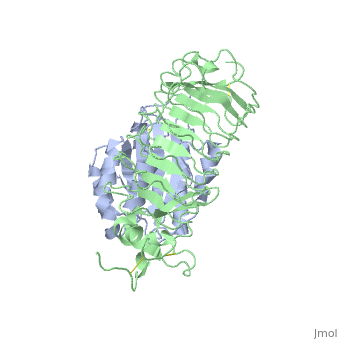
Crystal structure of the complex of Glycoprotein Ib alpha and the von Willebrand Factor A1 Domain
Structural highlights
DiseaseVWF_HUMAN Defects in VWF are the cause of von Willebrand disease type 1 (VWD1) [MIM:193400. A common hemorrhagic disorder due to defects in von Willebrand factor protein and resulting in impaired platelet aggregation. Von Willebrand disease type 1 is characterized by partial quantitative deficiency of circulating von Willebrand factor, that is otherwise structurally and functionally normal. Clinical manifestations are mucocutaneous bleeding, such as epistaxis and menorrhagia, and prolonged bleeding after surgery or trauma.[1] [2] Defects in VWF are the cause of von Willebrand disease type 2 (VWD2) [MIM:613554. A hemorrhagic disorder due to defects in von Willebrand factor protein and resulting in impaired platelet aggregation. Von Willebrand disease type 2 is characterized by qualitative deficiency and functional anomalies of von Willebrand factor. It is divided in different subtypes including 2A, 2B, 2M and 2N (Normandy variant). The mutant VWF protein in types 2A, 2B and 2M are defective in their platelet-dependent function, whereas the mutant protein in type 2N is defective in its ability to bind factor VIII. Clinical manifestations are mucocutaneous bleeding, such as epistaxis and menorrhagia, and prolonged bleeding after surgery or trauma. Defects in VWF are the cause of von Willebrand disease type 3 (VWD3) [MIM:277480. A severe hemorrhagic disorder due to a total or near total absence of von Willebrand factor in the plasma and cellular compartments, also leading to a profound deficiency of plasmatic factor VIII. Bleeding usually starts in infancy and can include epistaxis, recurrent mucocutaneous bleeding, excessive bleeding after minor trauma, and hemarthroses. FunctionVWF_HUMAN Important in the maintenance of hemostasis, it promotes adhesion of platelets to the sites of vascular injury by forming a molecular bridge between sub-endothelial collagen matrix and platelet-surface receptor complex GPIb-IX-V. Also acts as a chaperone for coagulation factor VIII, delivering it to the site of injury, stabilizing its heterodimeric structure and protecting it from premature clearance from plasma. Evolutionary Conservation Check, as determined by ConSurfDB. You may read the explanation of the method and the full data available from ConSurf. Publication Abstract from PubMedTransient interactions of platelet-receptor glycoprotein Ibalpha (GpIbalpha) and the plasma protein von Willebrand factor (VWF) reduce platelet velocity at sites of vascular damage and play a role in haemostasis and thrombosis. Here we present structures of the GpIbalpha amino-terminal domain and its complex with the VWF domain A1. In the complex, GpIbalpha wraps around one side of A1, providing two contact areas bridged by an area of solvated charge interaction. The structures explain the effects of gain-of-function mutations related to bleeding disorders and provide a model for shear-induced activation. These detailed insights into the initial interactions in platelet adhesion are relevant to the development of antithrombotic drugs. Structures of glycoprotein Ibalpha and its complex with von Willebrand factor A1 domain.,Huizinga EG, Tsuji S, Romijn RA, Schiphorst ME, de Groot PG, Sixma JJ, Gros P Science. 2002 Aug 16;297(5584):1176-9. PMID:12183630[3] From MEDLINE®/PubMed®, a database of the U.S. National Library of Medicine. See AlsoReferences
| ||||||||||||||||||
Categories: Homo sapiens | Large Structures | Gros P | Huizinga EG | Romijn RAP | Schiphorst ME | Sixma JJ | Tsuji S | De Groot PG
