Introduction
The protein gluten is found in wheat and grains such as rye and barley. Gluten is also involved with inducing an inflammatory response in individuals with celiac disease. Individuals who have the disease cannot digest gluten due to the structure of the protein, which will damage the small intestine. If an individual with celiac disease ingests foods containing gluten, the immune system responds by damaging the villi, which are fingerlike projections lining the small intestine. This immune response reduces the body’s ability to absorb nutrients that pass through the small intestine and into the bloodstream. As a result of the damaged villi, people with celiac disease can become malnourished. Although celiac disease is genetic, the research reviewed for this study placed emphasis on how the protein triggers an immune response in the gastrointestinal tract of affected individuals.
Gluten is a protein complex comprised of 2 components: gliadin (the water-soluble component) and glutenin (the water-isoluble component). Gliadins, for those with celiac disease, are the principle toxic component of gluten and are composed of proline and glutamine-rich peptide sequences. The peptides enter the circulatory system and come into contact with lymphocytes and T-cells, resulting in the release of cytokines. The cytokines interact with the villi of the small intestine and damage them, disabling the body from nutrient absorption. The symptoms can include abdominal pain, weight loss, fatigue, and many other symptoms associated with malnutrition. As of now, the only treatment for celiac disease is the total exclusion of gluten from the person’s diet.[1][2][3]
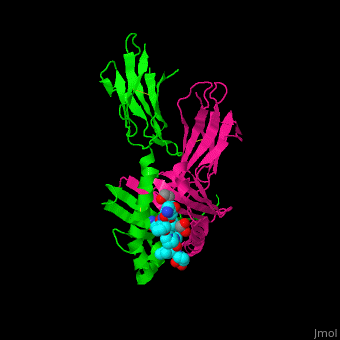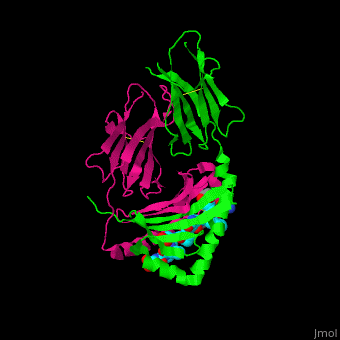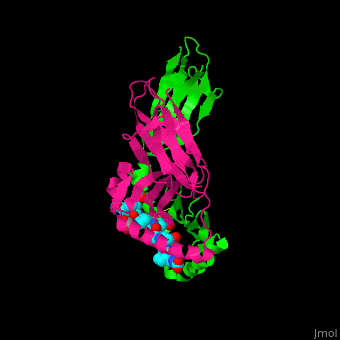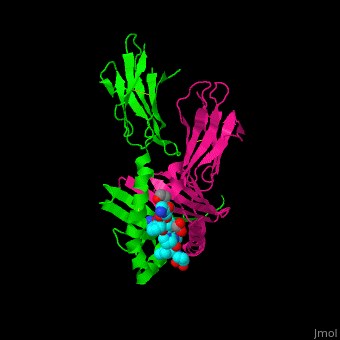
(PDB code 1s9v).
Gluten Protein Complex
Function
The gluten protein complex is made up of gliadin and glutenin components. Of the complex, gliadin directly affects the induction of an innate immune response via the . In the small intestine of patients with celiac disease, HLA-DQ2 restricted T-cells are present. After ingestion of a gluten product, the gliadin peptides enter the circulatory system and come into contact with lymphocytes and the gliadin-specific,, which is the fundamental step in producing the inflammatory response associated with celiac disease.[4]
Relevance
Human leukocyte antigens (HLA) are responsible for regulation of the immune system. The binding of gliadin peptides to HLA should be the same in celiac and non-celiac patients. However, it is unclear why only specific individuals produce the gliadin-specific, HLA-DQ2 restricted T-cells with pathogenic consequences. These implications suggest an underlying genetic component. [4]
Immune Response
HLA-DQ2 and HLA-DQ8
HLA-DQ2 and proteins occur at 95% and 5% respectively in all patients with celiac disease. The proteins HLA-DQ2 and HLA-DQ8 amplify the autoimmune response by binding the gluten complex to the transglutaminase in the tissue of the small intestine lumen. The new complexes are comprised of three chains: two MHC class II antigens composed largely of alpha helical and beta sheet structures and a gluten peptide. The MHC class II molecules HLA-DQ2 and HLA-DQ8 are human leukocyte antigens associated with the genetic risk of developing celiac disease and serves as a MHC class II molecule in the immune system of the body. In addition, the complexes have conformations that only expose the gliadin sequence that has gastrointestinal protease resistance. Since the conformation only exposes the resistant sequence, the body is unable to break down the complex. As a result, the body sends out antibodies, which bind the epitopes of the complex thus labeling it as a toxin. The end result is an amplified autoimmune response that attacks the lining of the small intestine to help rid the body of the resistant complex.[5]
Interactions
The gluten protein complex binds the HLA-DQ2 . Glutamine and lysine hydrogen bond to water, the nitrogen backbone of gliadin, and the asparagine, lysine, tyrosine and serine hydrogens of HLA-DQ2. The proline amino acids are all exposed to the external environment and do not participate in hydrogen binding while also preventing other amino acids from hydrogen binding through steric hindrance. Only proline-rich complexes are able to hydrogen bond properly with HLA-DQ2. All other proteins do not have the proper conformation and organization of proline to effectively bind HLA-DQ2, making the HLA-DQ2-gliadin complex very specific.[6]
When the gluten peptide enters the HLA-DQ8 antigen binding cleft and makes contact, .[7] Notable contacts include: 16 direct hydrogen bonds, 24 water mediated hydrogen bonds, and four salt bridges. The side chains of two glutamic acids and one phenylalanine buried deep in pockets of the complex serve to anchor the peptide within the antigen-binding groove. A proline and serine can be found in shallow pockets with their side chain serving as minor anchor residues to the peptide.
Treatments
Prolyl endopeptidases (PEPs) are a family of Serine proteases that help accelerate the breakdown of peptides by cleaving after proline residues. Since gluten is a proline-rich complex, these enzymes are being explored as a therapy to help treat individuals with celiac disease, since these individuals aren’t able to break down gluten naturally. [8]
|
|
|
Image Copyright 2005, National Academy of Sciences[9].
|
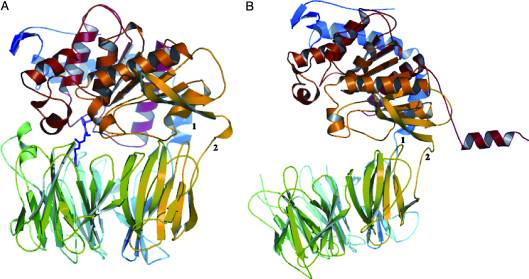
A study was done to explore the structural features of two bacterial PEPs with the purpose of evaluating their ability to cleave proline-rich residues. One was isolated from Myxococcus xanthuswith a and one was isolated from Sphingomonas capsulata in an . Both PEPs have two domains: a catalytic binding site that forms interactions with the substrate and the propeller domain that acts as a sort of stabilizing clamp structure. [8]
|
|
|
Image Copyright 2005, National Academy of Sciences[9].
|
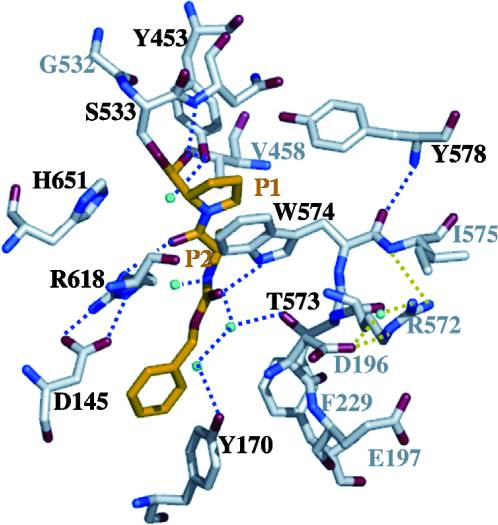
With further investigation of domain features of the PEP isolated from Myxococcus xanthus, interactions were observed that give the enzyme its proline-cleaving properties. Interactions between the domains of the PEP isolated from Myxococcus xanthus and the inhibitor in the binding pocket, which is colored yellow. Residues from the catalytic domain are labeled in black with their hydrogen bonds represented by blue dashed lines and residues from the propeller domain are labeled in gray with the salt bridges represented by yellow dashed lines. [8]
Using the structural components of the open and bound forms of PEP enzymes, a mechanism was proposed in which the incoming proline-rich peptide causes a conformational change that opens the catalytic binding site. This conformation is stabilized by the prolines in the substrate interacting with the arginine and aspartate residues in the binding site. The propeller region does not interact with the bound substrate, but the aspartate and glutamate residues interact with arginine residues in the catalytic region to stabilize the unbound form of the enzyme. [8]
Bacterial PEPs may be able to detoxify immunotoxic proline-rich peptides in the gut lumen of celiac patients by breaking down the gliadin before it reaches the small intestine. Further research would need to be done on how these enzymes can be made more acid-stable to withstand the acidic environment of the human intestine. Once these modifications are made, bacterial PEPs can be given orally as a therapeutic treatment for those with celiac disease. [8]
Studies have been done to determine the feasibility of the therapeutic implementation of bacterial PEPs for detoxification of gliadin complexes in individuals with celiac disease. The study showed that a substantially high concentration of PEPs as well as long exposure times (3 hours) were required for a complete detoxification of gliadin peptides and thus prevent intestinal transport of the peptides. [10]