Automated computational design of human enzymes for high bacterial expression and stability
Adi Goldenzweig, Moshe Goldsmith, Shannon E Hill, Or Gertman, Paola Laurino, Yacov Ashani, Orly Dym, Tamar Unger, Shira Albeck, Jaime Prilusky, Raquel L Lieberman, Amir Aharoni, Israel Silman, Joel L Sussman, Dan S Tawfik and Sarel J Fleishman [1]
Molecular Tour
Upon heterologous overexpression, many proteins misfold or aggregate, thus resulting in low functional yields. Human acetylcholinesterase (hAChE), an enzyme mediating synaptic transmission, is a typical case of a human protein that necessitates mammalian systems to obtain functional expression. Using a novel computational strategy, an AChE variant containing 51 mutations was designed that improved core packing, surface polarity, and backbone rigidity. This variant expressed at ~2,000-fold higher levels in E. coli compared to wild-type hAChE, and exhibited 20°C higher thermostability with no change in enzymatic properties or in the active-site configuration as determined by crystallography. To demonstrate broad utility, similarly, four other human and bacterial proteins were designed. Testing at most three designs per protein, enhanced stability and/or higher yields of soluble protein in E. coli were obtained. The algorithm requires only:
- A 3D structure of the protein (either experimentally determined or a high-quality model)
- Several dozen sequences of naturally occurring homologs
The algorithm, PROSS (Protein Repair One-Stop Shop), is available at http://pross.weizmann.ac.il .
. Wild type hAChE (PDB entry: 4ey4) is shown in blue and 51 mutated positions, which are distributed throughout dAChE4, are indicated by orange spheres.
The choice of mutations at Gly416 in hAChE illustrates the role of the two filters,
- a sequence alignment scan
- a computational mutation scan
that are used in pruning false positives (see the static image below). Position 416 is located on a partially exposed helical surface, where the small and flexible amino acid Gly is likely to destabilize hAChE. Indeed, in the alignment of AChE homologs, Gly appears infrequently and His is the most prevalent amino acid. Modeling shows, however, that in this specific context of hAChE, His adopts a strained side-chain conformation; in contrast, Gln, the third most prevalent amino acid, is predicted to be most stabilizing owing to its high helical propensity and favorable hydrogen-bonding with Tyr504. The combined filter, therefore, favors Gln over His for downstream design calculations.
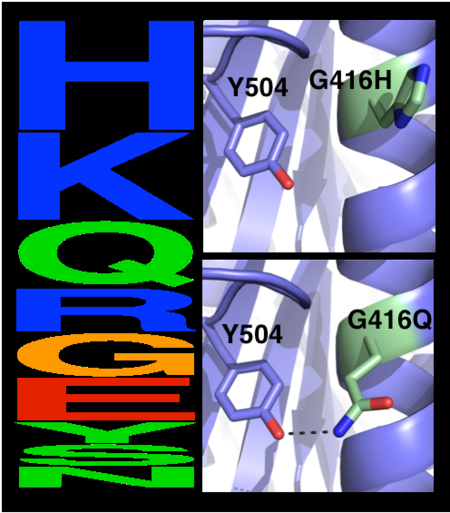
Eliminating potentially destabilizing mutations through homologous-sequence analysis and computational mutation scanning. Left: Sequence logo for hAChE position Gly416. The height of letters represents the respective amino acid’s frequency in an alignment of homologous AChE sequences. The evolutionarily ‘allowed’ sequence space (PSSM scores ≥0) at position 416 includes the 9 amino acids shown. Right: Structural models of mutations to the evolutionarily favored amino acid His, and to Gln, which is favored by Rosetta energy calculations. The His side chain is strained due to its proximity to the bulky Tyr504 aromatic ring, whereas the Gln side chain is relaxed and forms a favorable hydrogen bond with Tyr504 (dashed line)
Scenes highlight stabilizing effects of (in red), dAChE4 (PDB entry: 5hq3, green) compared to hAChE (PDB entry: 4ey4, cyan):
in the crystallographic structure of dAChE4 (PDB entry: 5hq3, green) compared to hAChE (PDB entry: 4ey4, cyan).
.
Comparison of the dAChE4 design model (yellow) with the solved crystal structure (PDB entry: 5hq3, green) and (wild-type hAChE (PDB entry: 4ey4, cyan):
- .
- .
- in the model and structure is 3.1 Å (dashed line). This conformation change likely results from elimination of a side chain-backbone hydrogen bond between Thr112 and Ser110 due to the designed Thr112Ala mutation.
- . Val331Asn was predicted to form a hydrogen bond with Glu450 and another with Pro446 in the designed model; in the crystal structure, instead, Asn331 interacts with Glu334 and Glu450.
- .
PDB reference: Stable, high-expression variant of human acetylcholinesterase, 5hq3.


