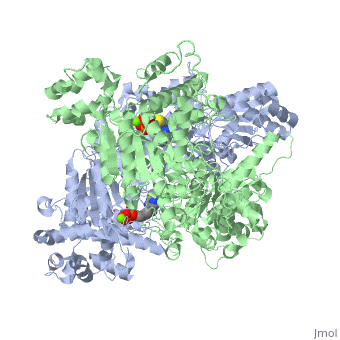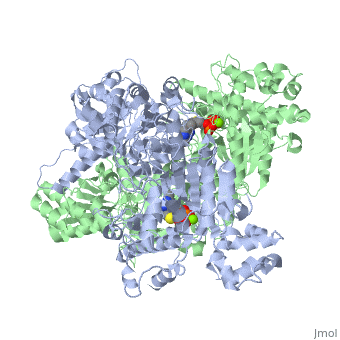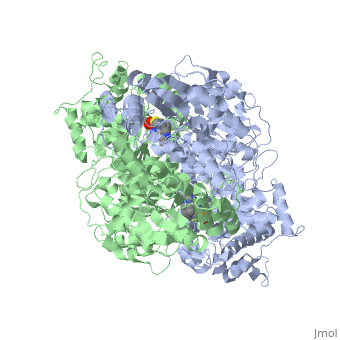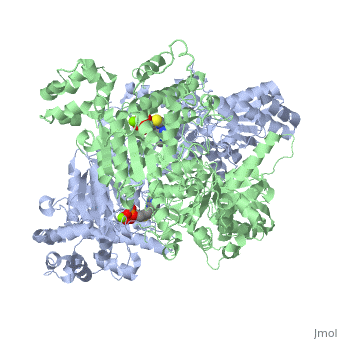
Function
Pyruvate dehydrogenase (E1) is one of three main components of the multienzyme complex pyruvate dehydrogenase. It accompanies dihydrolipoyl transacetylase (E2) and dihydrolipoyl dehydrogenase (E3) in comprising this multienzyme complex. The E. coli enzyme complex has a weight of approximately 4600-kD and a diameter of about 300 angstroms. The core of the particle is made of 24 E2 proteins arranged in a cube, which is surrounded by 24 E1 proteins and 12 E3 proteins. Together, these enzymes are responsible for synthesizing acetyl-CoA from pyruvate just prior to entrance into the citric acid cycle. Therefore, pyruvate dehydrogenase contributes to linking the glycolysis metabolic pathway to the citric acid pathway. For more details on E3 see Dihydrolipoamide dehydrogenase.
See also Pyruvate Dehydrogenase (Hebrew).
Structure
Pyruvate dehydrogenase (E1) falls within the class of alpha and beta proteins[1], containing . It is a multimeric protein. Mammalian E1s, including human E1, are heterotetrameric, composed of two α- and two β- subunits[2]. E1 from E. coli is, however, a homodimer with a molecular weight of 99474 containing α/β folds. It has two catalytic sites located at the interface between subunits. Each polypeptide chain consists of 886 residues[3]. The structure shown is the E. coli E1 pyruvate dehydrogenase component, PDB code
1l8a[4].
Reactions and Mecahanism
The multienzyme complex together catalyzes five distinct reactions in the conversion of pyruvate to acetyl-CoA. The overall result is described by the following reaction:
Pyruvate + CoA + NAD+ ==> Acetyl-CoA + CO2 + NADH
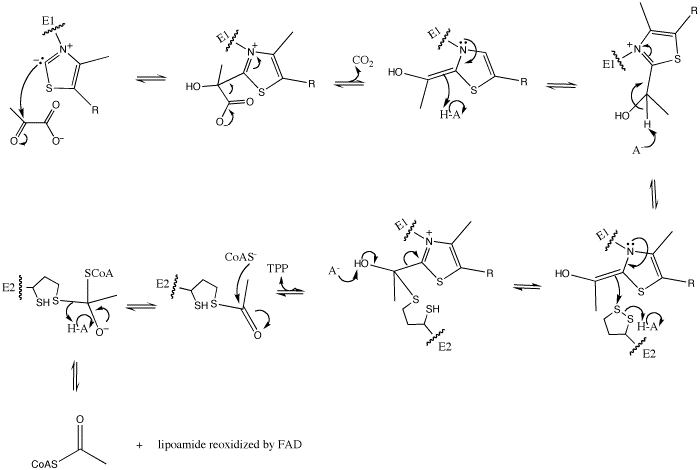
However, pyruvate dehydrogenase (E1) is responsible for only the first two of the five reactions. The first of these is the decarboxylation of pyruvate and coupling of thiamine pyrophosphate (TPP) to form hydroxyethyl-TPP.
Pyruvate + TPP ==> Hydroxyethyl-TPP + CO2
The enzyme requires as cofactors for catalysis.
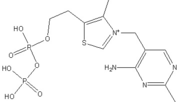
Thiamine pyrophosphate (TPP) E1 cofactor.
In this reaction, the ylide form of TPP attacks the electrophilic carbonyl group of pyruvate. This reflects the ability of TPP’s thiazolium ring, which primarily interacts with , to add to carbonyl groups. Decarboxylation of the resulting alkoxide yields an enol complex. This enol resonates to form the ylide form of hydroxyethyl-TPP[5]. During this reaction, the catalytic Mg2+ ion coordinates octahedrally with three protein ligands: , which bind TPP, and [3].
The second E1-catalyzed reaction is the transfer of the hydroxyethyl group to the lipoamide group of the next enzyme, dihydrolipoyltransacetylase (E2). The E2 lipoamide group consisting of lipoic acid linked to the amide group of a Lys residue. Lipoic acid contains a reactive cyclic disulfide that is reversibly reduced to give dihydrolipoamide. The ylide form of the hydroxyethyl group of the hydroxyethyl-TPP complex attacks this disulfide bond. TPP is then eliminated as it detaches with E1 and subsequently binds to the next pyruvate molecule[5].
E1-Hydroxyethyl-TPP + E2-Lipoamide ==> E1-TPP + Acetyl-dihydrolipoamide-E2
The overall mechanism is shown as following:
Regulation
Pyruvate dehydrogenase plays a key role in the regulation of the Krebs cycle. The reactions catalyzed by the pyruvate dehydrogenase complex constitute the only biological pathway for acetyl-CoA synthesis from pyruvate. It is thus crucial that these reactions be precisely controlled.
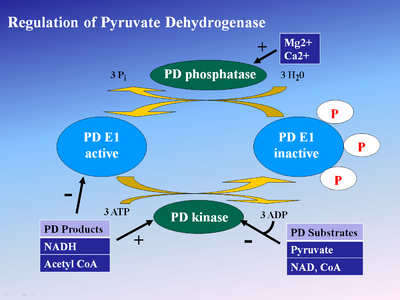
One method of regulation is product inhibition by NADH and acetyl-CoA. NADH is a product of reactions catalyzed by dihydrolipoyl dehydrogenase (E3), while acetyl CoA is a product of the aforementioned dihydrolipoyl transacetylase (E2). Both compounds compete for active sites on their respective enzymes. NADH competes with NAD+ for E3 active site, while acetyl-CoA competes with CoA for E2 active site. High NADH levels keep E3 in its reduced form, thus the E2 lipoamide group stays in its reduced state. This in turn prevents E1 from transferring the hydroxyethyl group to E2. Consequently, E1 activity is reduced. Similarly, Acetyl CoA reduces E1 activity by occupying binding sites so less pyruvate binds to E1. Thus, high relative NADH and Acetyl-CoA concentrations regulate E1 activity through product inhibition.
These two compounds also activate the pyruvate dehydrogenase kinase associated with the enzyme complex[5]. This results in phosphorylation of three different E1 serine residues (Ser 203, Ser 264, Ser 271) in human E1 and enzyme inactivation[6]. Enzyme regulation through phosphorylation by pyruvate dehydrogenase kinase and dephosphorylation pyruvate dehydrogenase phosphatase has been implicated as a target for treating cancer, heart ischemia, and diabetes[7].
Kinetics
E. coli pyruvate dehydrogenase binding constant and maximum velocity values have been reported as Km = 0.3 mM and Vmax = 5,500 kat/mol (37 degrees C, pH = 7.6, 5 microM pyruvate, and 3 mg/L protein). The multienzyme complex exhibits postive cooperative binding (Hill constant = 1.9)[8].
Medical Implications
Pyruvate dehydrogenase is an autoantigen recognized in primary biliary cirrhosis. These antibodies appear to recognize oxidized protein that has resulted from inflammatory immune responses. Some of these inflammatory responses are explained by gluten sensitivity[9]. Other mitochondrial autoantigens include oxoglutarate dehydrogenase and branched-chain alpha-keto acid dehydrogenase complex, which are antigens recognized by anti-mitochondrial antibodies.
Pyruvate dehydrogenase (PDH) deficiency is a congenital degenerative metabolic disease resulting from a mutation of the pyruvate dehydrogenase complex (PDC) located on the X chromosome. Although defects have been identified in all 3 enzymes of the complex, the E1-α subunit is predominantly the culprit. Malfunction of the citric acid cycle due to PDH deficiency deprives the body of energy and leads to an abnormal buildup of lactate. PDH deficiency is a common cause of lactic acidosis in newborns and often presents with severe lethargy, poor feeding, tachypnea, and cases of death have occurred[10].
3D structures of pyruvate dehydrogenase
Pyruvate dehydrogenase 3D structures