The human Factor VIII, also known as anti-hemophilic factor (AHF), is an essential blood-clotting protein [1]. It consists of 2332 residues [2]. Its gene is located on the X chromosome [1][3].
Factor VIII is produced inside the liver (by the sinusoidal cells) and outside (by the endothelial cells) and acts in the intrinsic pathway of blood coagulation [1]. It is actually the lack or the deficiency of the factor VIII (which is a plasma glycoprotein) that causes a bleeding disorder: hemophilia A [2].
Factor VIII is much studied in order to find a cure for hemophilia A (also written as HEMA), for instance by designing mimicking factors [4].
History
1937: first use of the factor VIII (known as “Antihemophilic Globulin”) to cure blood coagulation disorders thanks to the discovery of F.H.L Patek and A.J Taylor [5].
1964: Usual utilisation of concentrated factor VIII to treat hemophilia [6].
1984: Factor VIII was first characterized by scientists at Genentech [7].
2017: Concentrated factor VIII with extended half-life [8].
Function
Factor VIII plays a central role in blood coagulation
The Factor VIII circulates in the bloodstream in its inactive form, bound to another molecule called Von Willebrand Factor, until an injury that damages blood vessels occurs. In plasma, factor VIII exists predominantly in a complex with the Von Willebrand factor, because this latter plays a role of stabilization. By contrast, in its free state, factor VIII is rapidly cleaved by Serine Proteases [1][7].
The coagulation process
In response to an injury, the coagulation factor VIII is separated from von Willebrand factor. The active form (called “Factor VIIIa”) is obtained by a proteolytic cleavage of the B-domain of Factor VIII by Thrombin [1][2]. Then the two remaining chains are linked together thanks to a metal link (probably calcium ion) [2].
Thus the factor VIIIa is a non-covalent dimer [2].
It is the catalyst for the activation reaction of the factor X (to Factor Xa) by activated Factor IXa in the presence of calcium ion and phospholipids.
This activation reaction is accelerated approximately 200,000 times when factor VIII is present. [1][2][7]
Then, no longer protected by the von Willebrand factor, the factor VIIIa is proteolytically inactivated and quickly cleared from the blood stream, whereas, factor Xa becomes able (with the help of other factors) to stop the bleeding by forming a blood clot. [1][7]
Structure
Primary Structure
In humans, factor VIII is encoded by the F8 gene [2][9][10]. This gene maps on the most distant band of the long arm of the X-chromosome (region Xq28). It is 186 kb in size (0.1 % of the whole size of the chromosome) and contains 26 exons [3].
Secondary Structure
Factor VIII protein is composed of six globular domains: A1-A2-B-A3-C1-C2 and contains one Ca2+ and two Cu2+ ions. It has a molecular weight of 330 kDa [2][7][9].
In the following, the structure of an engineered protein is further described. This protein has no B domain to mimic the active factor VIIIa. In addition, it is more amenable to structural studies because it shows higher stability expression levels and structural homogenity [2][11].
The three A domains are homologous to the A domains of the copper-binding protein Ceruloplasmin [1][7]. Together, they form a triangular heterotrimer where the A1 and A3 domains interact with the C2 and C1 domains, respectively [2].
The C domains belong to the phospholipid-binding discoidin domain family [1]. They are adjacent at the base of the triangular heterotrimer. Moreover, C1 and C2 domains are structurally homologous and they have the ability to bind the membrane. Indeed, both C domain protrude three β-hairpin loops with hydrophobic and basic residues in the same direction. Thanks to these loops the factor VIII might interact with the phospholipid bilayer. [2]
Factor VIIIa is obtained by cleavage and release of the B domain [1][2][11]. Although factor VIIIa can be formed from at least two cleavages involving Arg372 and Arg1689, fully active factor VIIIa is obtained only after a third cleavage at Arg740 [2].
The two chain that result are a heavy and a light chains [2][7][10].
• The has a various size (90 or 120 kDa) [1][12]. It consists of the A1 and A2 domains [1][7][12]. Both domains are built up of two connected β barrels [2].
• The has a molecular weight of 80 kDa and is composed of 684 amino acids [12]. It contains two domains: a unique A domain of 371 amino acids and a duplicated C domain of 153 amino acids and 160 amino acids, respectively [12]. These domains are ranked in the following order A3-C1-C2 [1][12]. It is composed of 42 % irregular structure, 36 % β-strands, and 22 % α-helices [12]. The C1 and C2 domains are defined by a distorted β barrel, while A3, as well as A1 and A2, is composed of two connected β barrels [2]. This chain also contains of the major binding site of von Willebrand Factor at its N-terminus [12].
Figure 1: Domain organisation of the uncleaved coagulation factor VIII (top) and the engineered factor without B domain (bottom). (Figure adapted from [2])
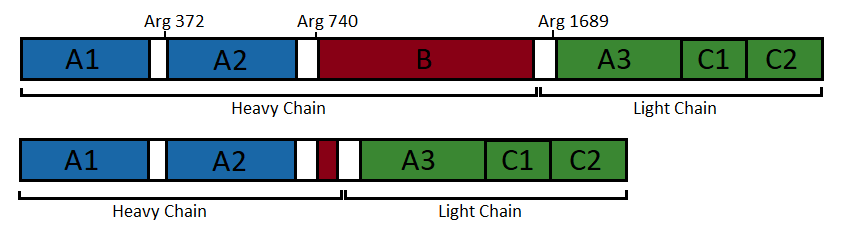
Both chains are non-covalently associated through to a calcium ion to form the active heterodimer [2][12]. This complex is the pro-coagulant factor VIIIa [1].
Such an association is essential for the functioning of the factor VIII [12].
Ligands
The calcium ion (Ca2+) and the copper ion (Cu2+) are both ligands the factor VIII is able to bind to [10]. More precisely, in factor VIII there are two copper ions and their binding sites are located internally within the and the domain. The domain binds another ligand, a , bound to its own binding site. [2]
One other molecule can be found on this protein: N-acetyl-D-glucosamine. This molecule is covalently bound to Asn residues of the protein during the maturation process in the endoplasmic reticulum and the Golgi apparatus [13]. N-acetyl-D-glucosamine is not a ligand since it is not a specific substrate that binds a specific site in the protein. Indeed many proteins have such a glycolysation on their asparagine residues [14]. N-acetyl-D-glucosamine is a post-translational modifications and may be different depending on the physiological context [15][16].
The alpha-D-mannose molecule, present in the structure shown here, might also be a posttranslational modification, since it is a sugar that can be establish N-type bonds[17]. Factor VIII is thus a glycoprotein [14].
Disease
Hemophilia is a genetic disorder characterized by a permanent tendency to hemorrhage because of a lack of blood coagulation [2].
There are different types of hemophilia: A or B, caused by a deficiency of two different factors.
Hemophilia A (HEMA), is four times as common as hemophilia B.
It is caused by a deficiency of factor VIII. [18]
This deficiency in factor VIII clotting activity results in prolonged delayed or recurrent bleeding prior to complete wound healing [19].
Although hemophilia A is usually an inherited disease and therefore runs in families [18], about one-third of people with the disease are caused by a spontaneous mutation [18] such as misense or nonsense mutations, gene deletions or inversions [2].
Hemophilia A can be mild, moderate, or severe, depending on the level of Factor VIII clotting activity [19][20].
The major treatment of the bleeding disorder associated with hemophilia A is the infusion of factor VIII, which leads to the correction of hemostasis [18].
Inheritance
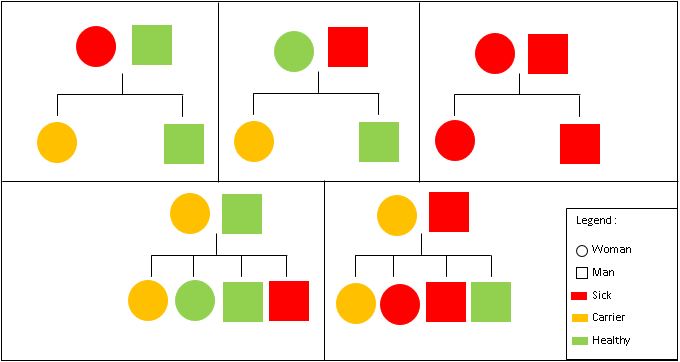
Hemophilia A is inherited in an X-linked recessive manner. This means that if a son inherits an X chromosome carrying hemophilia from his mother, he will have hemophilia. By contrast, daughters, even if they inherit one hemophilia allele, can compensate it with their second healthy X chromosome. As a result, women only rarely have symptoms, but women that are carriers, may pass the gene on to their children (50% chance per pregnancy) [19].
Figure 2: Hemophilia Inheritance
Relevance
Hemophilia occurs in approximately 1 in 5,000 live births but it is severe in approximately 60% of cases.
The main medication to treat hemophilia A is concentrated factor VIII protein, called “clotting factor”. Getting this “clotting factor” is therefore a major concern for hemophilia-affected people [18].
Nowadays, recombinant coagulation factor VIII products are developed in labs through the use of DNA technology [7][11]. For instance, Toole and colleagues have created a biologically fully active factor with improved heterologous expression efficiency by deleting the B-domain from the native human factor VIII [11].


