Introduction
(SMP) holophosphatase complex functions as a key regulator of the receptor tyrosine kinase (RTK) signaling pathway by removing an inhibitory phosphate on the RAF family of proteins to allow for MAPK signaling.[1] This interaction of the RTK-RAS pathway and the SMP complex drives cell proliferation.[2] The SMP complex is made of three subunits, SHOC2, PP1C, and MRAS. Each of these subunits has a different shape that corresponds to its different function. uses a crescent shape to enhance substrate interactions and complex stability.[3] contains the the catalytic site of the complex which dephosphorylates the N-terminal phosphoserine (NTpS) of RAF.[3] binds to GTP which causes assembly of the SMP complex. The localizes the complex to the cell membrane.[3] Once the SMP compelx is assembled, MRAS can bind to , allowing the signaling cascade to continue. Mutations in one or multiple of these subunits can lead to over-activation of the signaling pathway, which may result in cancer and developmental disorders called RASopathies.[1]
There are many regulatory mechanisms that serve as a lock on this RAS-MAPK pathway, decreasing the likelihood of unintentional pathway activation. [2] One example is , a protein dimer that keeps inactive RAF localized to the cytoplasm. An (NTpS) keeps RAF bound to this protein dimer, and when the SMP complex is assembled, the catalytic subunit, PP1C, removes the phosphate group from Ser259, releasing RAF from , and activating the RAS-MAPK cell proliferation pathway. [2]
In all images and animations, SHOC2 will be shown as cyan blue, MRAS as lime, and PP1C as violet. Other important components involved in the function of the SMP complex include the 14-3-3 dimer and Raf, which will be shown in salmon and slate-blue, respectively.
SMP Complex Mechanism
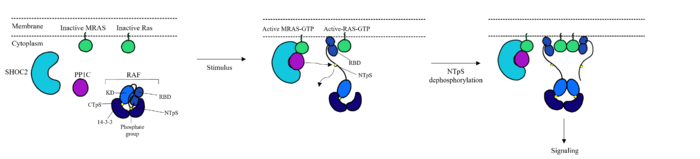
Figure 1: Mechanism of SMP complex formation and activation of RAF.
[3][4] The RAS-RAF signaling cascade is inhibited when RAF is phosphorylated at Ser259.[1] There is a dimer present in the cytoplasm that interacts with RAF through hydrogen bonds between R129 of 14-3-3 and Ser259 of RAF when Ser259 is phosphorylated. This interaction causes an as 14-3-3 restricts RAF to the cytoplasm and sterically inhibits RAF from binding with RAS. This interaction is crucial in regulating cell proliferation, as it prevents cell growth in the absence of a signal. Extracellular Growth Factors cause GTP to bind to MRAS which triggers SMP formation [4].Upon SMP complex formation, PP1C is brought into close proximity of RAS, leading to the dephosphorylation of Ser259 of RAF by the active site of PP1C [4]. Once dephosphorylated, RAF is in the
, allowing RAS to bind RAF, initiating the signaling cascade.[5]
Structure of Subunits
SHOC2
is essential for complex formation. It is a crescent shaped complex that serves as a bridge for PP1C and MRAS, maximizing interaction between the three subunits of the SMP complex [2]. SHOC2 contains a large leucine rich region (LRR) that provides stability and localizes subunit PP1C to the membrane[3]. SHOC2 only undergoes a when PP1C and MRAS bind showing it is a scaffolding protein that provides a favorable interface for complex formation[3]. SHOC2 depletion is being studied as a therapeutic approach for RAS-driven cancers due to large scale interactions of the subunits being made possible by SHOC2 [1]. As shown in Figure 1 SHOC2 and PP1C first engage in binding with each other via an N-terminal on SHOC2 that is complimentary to a binding sequence on PP1C. SHOC2 residues embed in the complimentary region of PP1C, enhancing SHOC2 affinity for PP1C. SHOC2 binds MRAS-GTP through β strands of a LRR that interacts with a hydrophobic region of MRAS-GTP further stabilizing the complex[1].
PP1C
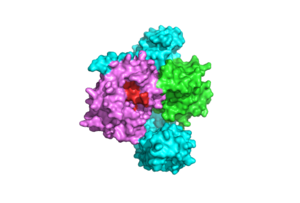
Figure 2: Catalytic Site of PP1C (PDB 7DSO). The catalytic site is shown in red. PP1C undergoes minimal conformational change when binding to SHOC2 and MRAS to ensure the active site is available for binding of RAF
[3].
subunit contains the catalytic site of the SMP complex. PP1C is a Phosphatase enzyme responsible for the removal of a phosphate group on the N-terminal phosphoserine (NTpS) of RAF (Ser259)[3]. The exact mechanism of dephosphorylation is currently unknown, but there are three catalytic metal ions: 2 Mn⁺² and 1 Cl⁻¹ present that coordinate nucleophilic water molecules in the active site [2]. This dephosphorylation event allows for pathway activation, as shown in Figure 1 [3]. Although PP1C can dephosphorylate other proteins independently from the SMP complex, it cannot act on RAF unless bound to the complex because it lacks intrinsic substrate selectivity [3]. SHOC2 and MRAS aid in the specificity of the enzymatic activity. PP1C binds to SHOC2 and MRAS-GTP in a specific orientation that doesn’t change the conformation of the catalytic site and leaves it accessible for substrate binding as shown in Figure 2.
PP1C binds to SHOC2 through a hydrophobic N-terminal disordered region that is complimentary to the and adjacent to a catalytic metal ions [3]. In the RAS/RAF signaling cascade, the region of RAF that is C-terminal to the phosphate group binds to this hydrophobic groove, and the remaining residues bind to the hydrophobic region of SHOC2 [2]. RAF binding to this region of SHOC2 is what allows PP1C to be specific when in the SMP complex in comparison to PP1C on its own [2]. Similarly to SHOC2, PP1C does not undergo a when SHOC2 and MRAS-GTP bind. The lack of conformational change shows that the structure of PP1C is not dependent on the SMP complex, but in order to act as a phosphatase it must be bound to the complex [3].
PP1C is involved in many different cellular signaling pathways including protein synthesis, muscle contraction, and even carbohydrate metabolism[6]. In all these pathways, including the SMP pathway, PP1C does not exist as a monomer, it is present in holoenzyme form complex with one of two regulatory subunits ensuring there is no sporadic pathway activation [3].
RAS/RAF
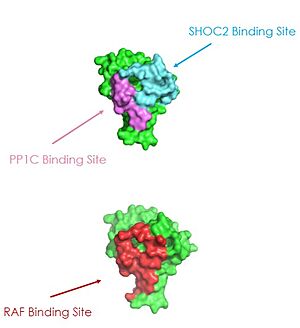
Figure 3: MRAS binding sites with SHOC2, PP1C, and RAF (PDB 7DSO)
[3].
RAF
While is not technically part of the SMP protein complex, it is crucial for advancement in the cell signaling pathway SMP helps mediate. RAF plays many different roles in this pathway and has many different domains. Figure 1 shows RAF has a RAS binding domain (RBD), a (NTpS), and a kinase domain[4]. Figure 1 also shows these domains and mechanistically how RAF is involved in signal advancement or lack thereof. When its N-terminal serine is phosphorylated RAF is bound to a 14-3-3 protein dimer, inactivating the pathway. As shown in Figure 1 the dephosphroylation of Ser259 starts the signaling cascade [4].
RAS
RAS proteins are GTP-dependent intracellular switches that are anchored to the plasma membrane. [3] RAS proteins activate RAF kinases through direct binding and membrane recruitment, resulting in RAF dimerization and pathway activation [3]. The SMP complex has specificity for MRAS. Other RAS proteins may bind to SHOC2, but MRAS induces the complex formation with a significantly lower dissociation constant [3]. There are no known membrane interacting regions on SHOC2 and PP1C, meaning the hydrophobic fatty acid tail on MRAS is responsible for recruiting the complex to the cell membrane. This allows only for 2D movement and increasing local concentrations of the players needed in this signaling pathway [2].
A significant amount of steric overlap is seen in MRAS for the binding sites of PP1C, SHOC2, and RAF [3]. In Figure 3, MRAS is shown in green, with the SHOC2 binding site colored cyan, the PP1C binding site colored violet, and the RAF binding site shown in red on a different RAS protein. Hence, multiple RAS proteins are required for further activation of the receptor tyrosine kinase pathway [4]. Due to the significant overlap in binding domains, one MRAS molecule is needed to recruit SHOC2 and PP1C to the membrane, and another RAS molecule is needed activate RAF [4]. The ability of MRAS-GTP to cluster at the cell membrane is a crucial capability for this protein complex. The presence of this is responsible for this anchoring to the cell membrane, similar to the hydrophobic fatty acid tail on MRAS that is responsible for recruiting SMP to the cell membrane.
MRAS contains two regions called Switch I (SWI) and Switch II (SWII) that undergo conformational changes depending if MRAS is bound to GDP or GTP [3]. The conformation of these switches determines if the SMP complex can form or not. Mutations to MRAS can lead to consistent GTP-loading, causing an increase in the formation of the SMP complex as well as consistent activation of the cell-proliferation pathway in the absence of external growth factors.
Switch I and Switch II
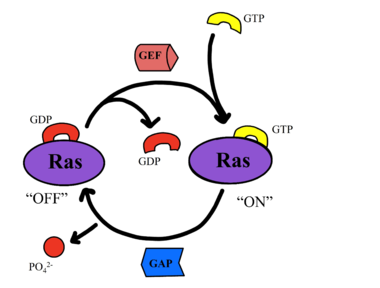
Figure 4: Exchange of GTP for GDP via nucleotide exchange factors
[3].
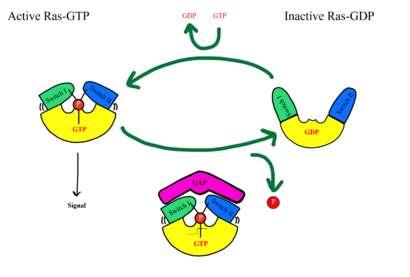
Figure 5: MRAS SWI and SWII open and closed conformations
[3].
SHOC2-PP1C-MRAS is a central gatekeeper in receptor tyrosine kinase signaling [3]. Figure 1 shows the specific pathways SHOC2-PP1C-MRAS mediates. When MRAS is bound to GDP, shown in the left of Figure 1, RAF is bound to a 14-3-3 protein dimer restricting it to the cytoplasm. When MRAS-GDP is exchanged for GTP via a nucleotide exchange factor GEF, shown in Figure 4, a conformational change occurs. This change causes a shift from the to of Switch I, shown in Figure 5. The Switch I (SWI) region is made up of [1]. These residues are crucial for the binding of MRAS, SHOC2, and PP1C because MRAS undergoes a conformational change that allows for SMP complex assembly upon GTP binding [2]. When GTP is bound to MRAS, it is in the “closed conformation” because hydrogen bond interactions between the γ phosphate of GTP and residues in the SWI region of MRAS cause SWI to adopt a closed conformation [2], as seen in Figure 5. The closed conformation allows for the binding of SHOC2 and PP1C because there is no steric clash between the and the surface of SHOC2 when GTP is bound [1]. The only large-scale conformational change occurs in the MRAS subunit [3]. When GDP is bound to the MRAS domain, it is in the “open” conformation. Since the γ-phosphate is not bound to GDP, there are no hydrogen bond interactions with the oxygens of the γ-phosphate group and the MRAS SWI region, causing MRAS to adpot an "open" conformation. Since SHOC2 and PP1C do not undergo much conformational change, they are in a slow equilibrium of binding and unbinding until MRAS binds to GTP allowing MRAS to bind to SHOC2 and PP1C [3].
Cancer and Rasopathies
Common mutations in SHOC2 and PP1C lead to amino acid changes on the interaction surfaces, which can result in higher binding affinity.[4]The interface of SHOC2-PP1C is stabilized by the mutation because this creates a salt bridge with E116 of PP1C. This enhances the binding energy by -22.7 kcal/mol. Mutations to MRAS can result in consistent GTP-loading, increasing the formation of the SMP complex in the absence of external growth factors that are necessary for activation of the pathway in a healthy organism. The majority of wild type MRAS in cells are bound to GDP, whereas the MRAS with the Q71L mutation locked MRAS in the GTP bound state.[2] In MRAS, both show increased interaction with other effectors such as BRAF, CRAF, and AF6, consistent with gain-of-function mutations that activate MRAS, leading to GTP-loading.
Mutations in PP1C can trigger increased active site activity, increasing the RAF proteins that are active and available to bind to RAS. In patients with Noonan Syndrome, a disease in the RASopathy family, a point mutation of MRAS was identified, however the effects this has are unknown.[5] Universally, when this MAPK cascade is unregulated, cells are able to proliferate regardless of external signals, leading to cancer and/or RASopathies.





