NOTCH1 is a member of NOTCH family receptor proteins which consists of four members (NOTCH1-4). NOTCH proteins are evolutionarily highly conserved signalling proteins responsible for direct transduction of developmental signals at the cell surface into change in a transcriptional profile in the nucleus.
Structure and Function of NOTCH1
NOTCH receptors are class I transmembrane glycoproteins composed of an extracellular subunit and transmembrane and intracellular subunit, which interact via a specialised heterodimerization domain (HD). The extracellular subunit engages ligand via several EGF-like repeats and further contains three LIN-12/NOTCH repeats (LNR) which stabilise the dimerization domain by holding the two NOTCH subunits together. The transmembrane-intracellular subunit contains a short extracellular juxtamembrane peptide, transmembrane sequence and cytoplasmic domains including RAM domain, nuclear localization signals (NLS), a series of ankyrin repeats, glutamine-rich region (OPA) and C-terminal PEST domain which serves as a ligand-activated transcription factor [1].
Proteolytic Events During NOTCH1 Secretion and Signal Transduction
Furin-type Convertase Cleavage
NOTCH1 is posttranslationally modified by a proteolytic cleavage at S1 sites and reaches the plasma membrane as a heterodimer. Non-cleaved NOTCH1 is autoinhibited. Furin-type convertase is responsible for this process and cleaves NOTCH1 in at least two places: after R1633 and after R1664 [2]. Both residues are located in a loop exposed into the cytosol and lie approximately 100 and 70 amino acids external from the transmembrane region, respectively [2][3].
Additional Cleavages in Response to Receptor Activation
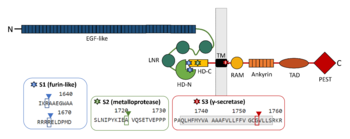
Schematic picture of NOTCH1 domains and cleavage sites with emphasis on regions contributing to the interaction between the two subunits.
Binding of NOTCH1 ligands such as Delta1 or Jagged1 leads to the dissociation of the heterodimer. This structural change reveals S2 site for a cleavage by metalloprotease TNAα-converting enzyme (TACE), a member of a disintegrin and metalloprotease domain (ADAM) family, which then produces a fragment termed as NOTCH extracellular truncation (NEXT). S2 site is located between A1710 and V1711 in murine NOTCH1, 13 amino acids from the TM domain
[4][5], which corresponds with positions 1720 and 1721 in human NOTCH1
[3]. The mechanism of ligand-induced dissociation can be explained by mechanical force caused by simultaneous endocytosis in the ligand cell. Thus, the events in the ligand cell are important for the NOTCH signal transduction as well. Since S2 cleavage is a ligand-regulated step, mutations in heterodimerization domain can mimic ligand-bound stage of the receptor and facilitate NOTCH proteolysis in a similar manner
[6]. The cleavage by metalloprotease probably brings the receptor in a conformation similar to that of constitutively active receptors
[4].
Although the cleavage at S2 site is prominent for the activation of the NOTCH pathway [4], subsequent cleavage by γ-secretase presenilin at S3 site is the one which is responsible for NOTCH intracellular domain (NICD) production [7]. γ-secretase cleaves murine NOTCH1 between G1743 and V1744 [8], which is G1753 and V1754 in human NOTCH1 [3]. The same enzymatic activity creates Aβ peptide in Alzheimer‘s disease from β-APP precursor [7].
Heterodimerization Domain and Negative Regulatory Region
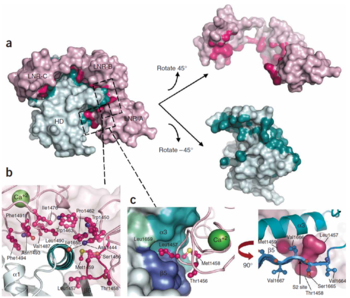
Interface between the LNR and HD domains. (A) LNR-HD contact interface. LNR domain surface coloured pink when the atom approaches within 4Å of the HD domain, otherwise coloured light pink. HD domain surface is coloured teal when the atom approaches within 4 A˚ of the LNR domain, otherwise light cyan. (B) Contacts with LNR including conserved hydrogen bond anchor helix 3 above S2 site. (C) The LNR-AB linker sterically blocks access to the S2 site. Hydrophobic pocket in HD domain houses the S2 site and residues from the LNR-AB linker (in ball-and-stick form).Three ‘gatekeeper residues’ from LNR-AB linker shown as a pink surface.
[9]Stable association of the two NOTCH1 chains depends on the heterodimerization domain which consists of two regions. 65 amino acid C-terminal region (HD-C, NTM) remains associated with the transmembrane part of the receptor, whereas 103 aminoacid extracellular N-terminal region (HD-N, NEC) has been separated by furin-type convertase and interacts with the membrane-bound part non-covalently. Adjacent to HD-N are three LIN-12/NOTCH repeats (LNR) which are not necessary for heterodimerization, but rather protect HD-C from metalloprotease cleavage and prevent ligand-independent activation of the signalling pathway. LNR are together with HD-N termed as the negative regulatory region, NRR
[10].
NOTCH mutants with deleted extracellular domain are constitutively active and are cleaved at the S3 site in a constitutive manner. Also, their activity is equivalent to NOTCH mutants containing the intracellular part only. This observation supports the idea that the conformation of the receptor is important for the changes in accessibility of S3 site. Ligand binding can change the conformation and permits cleavage of NOTCH by γ-secretase either directly or indirectly. On the contrary, constructs bearing also LNR are neither constitutively active nor constitutively cleaved [11].
T-cell Acute Lymphoblastic Leukaemia
T-cell-acute lymphoblastic leukemia (T-ALL) is an aggressive hematologic tumor that predominantly affects children and adolescents. T-ALL is more prevalent in males. T-ALL is manifested by malignant differentiation of the multipotent precursor cells of the lymphoid lineage which lost their ability to mature and became the leukemic blasts infiltrating bone marrow. T-ALL overall has a poor prognosis, 5-year relapse-free survival rate is over 75 % in children and 50 % in adults [12].
The importance of NOTCH1 mutations in T-ALL has been shown by Weng and colleagues [13]. 44 % of NOTCH-dependent T-ALL cell lines harboured gain-of-function mutations in the heterodimerization domain. Moreover, about 40 % of these cases were accompanied by mutations in the intracellular PEST domain of NOTCH1 which is important for the signal termination [14]. Weng and colleagues proposed a model of synergy in which mutations in the heterodimerization domain increase the production of NICD by γ-secretase cleavage while mutations in intracellular PEST domain increases its half-life. Cell lines with mutations in the heterodimerization domain are sensitive to γ-secretase inhibitors and exhibit G0/G1 cell cycle arrest opposed to PEST domain mutants which are unaffected by the inhibitor [13]. Common mutations found in human T-ALL and acting in γ-secretase-dependent manner are L1575P, L1594P, and L1601P [15][13].
Mutations in Heterodimerization Domain
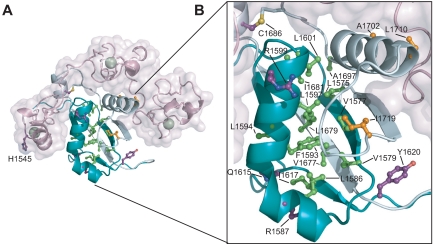
T-ALL tumor associated mutations in Notch1 NRR. (A) Structural representation highlighting T-ALL mutations. Side chains of residues mutated in T-ALL patients are shown in ball-and-stick form. Residues are colored according to mutation site: core (green), interface (orange), or partially exposed (purple). (B) Close-up view of mutations in the HD domain.
Activating mutations in the heterodimerization domain are predominantly missense mutations at sites which are conserved among vertebrate NOTCH receptors or small in-frame insertions and deletions. As a consequence, mutated receptors are activated regardless of the binding of a ligand [15][13]. The mutation hotspot is the region between residues 1574 and 1622 of the N-terminal portion of the heterodimerization domain which contains several highly conserved hydrophobic residues including a cluster of aliphatic amino acids. In this region are located activating mutations such as L1575P, L1594P, or L1601P which can increase the transcription from 3- to 9-fold [13]. However, some similar missense mutations in invariant amino acids can be also found in the C-terminal part of the heterodimerization domain [15][13]. Abovementioned mutations cause heterodimers to dissociate more readily. Among the mutations with the strongest effect on the heterodimer dissociation are L1601P, V1677D, and L1679P which induce dissociation of most or almost all furin-processed heterodimers into two subunits (the latter two mutations are in HD-C) [15]. The effect of these mutations can be explained from the structural properties of interacting chains which are forming antiparallel beta sheets followed by an alpha helix. Especially in case of a strong secondary structure breaker such as proline, this interaction can be impaired [16]. Besides that, NOTCH can be also activated by insertions. One mutation identified by Malecki and colleagues was a 16 amino acid long insertion which had negligible effect on heterodimer stability, but probably changed the relative position of S2 site in respect to the protective part of NOTCH [15].
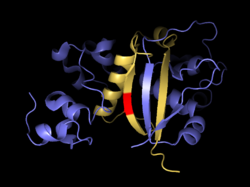
Antiparallel beta sheets formed by both subunits together. S2 site is in red.
Aberrant proteolytic processing can be also triggered by changes in spacing of the heterodimerization domain from the plasma membrane. These mutations are called juxtamembrane expansion (JME) alleles. These mutations are duplication insertions in the vicinity of the extracellular juxtamembrane region of the receptor and they depend more on the size of insertion than on the specific amino acid sequence. Insertions are clustered around position 1740 and range from 11 to 36 amino acids [17].
Some of the mutations such as L1601P and F1593S can affect furin cleavage. This might be caused by a retention of newly synthesised NOTCH in endoplasmic reticulum due to possible changes in folding, and recognition by ER-resident quality control mechanism [15].
Relevance
NOTCH1 Role in Normal T-cell Development
In hematopoietic system NOTCH1 plays a key role in T-cell development. NOTCH1 supports cell growth, proliferation and survival at multiple stages of thymocyte development. NOTCH1 activation drives multipotent progenitor cell towards T-cell lineage. NOTCH1 inhibition leads to ectopic differentiation of immature B-cells in thymus [18]. NOTCH1 is also involved in progression through double-negative stages in thymocytes development [19] and has an important role in rearrangement of T-cell receptors and decision whether γδTCR or αβTCR lineage will be developed [20].
NOTCH1 as a Therapeutic Target
The central role of NOTCH1 in T-cell signalling and high prevalence of NOTCH1 mutations in T-ALL cases led to targeting NOTCH1 in T-ALL therapy. The main approach to blocking NOTCH1 signalling is targeting γ-secretase that is essential for NOTCH1 signalling. γ-secreatase inhibitors (GSIs) reduce the production of Notch intracellular domain (NICD) and downregulate transcription of proteins promoting leukemic cell growth [21]. Antileukemic effects of GSIs were shown in vitro using γ-secretase inhibitor Compound E [22] and in vivo in a mouse model [23].
GSIs treatment as a side effect causes intestinal toxicity, because NOTCH1 plays an important role in intestinal stem cells. The side effects manifest in malabsorption and intestinal secretory cells metaplasia and accumulation of goblet cells [24].
It was shown in vitro that some T-ALL cases are resistant to NOTCH1 inhibition with GSIs. This is in some cases associated with constitutive activation of the PI3K-PTEN pathway [25] or with further mutations, such as FBXW7 (mutation in F-box protein) [26].
More recent approach uses specific antibodies against NOTCH proteins.These antibodies bind to the HD domain of NOTCH and thus inhibit signalization. The inhibition effect on tumor growth was shown in vitro and in vivo [27][28].
Structural Highlights
Majority of T-ALL-associated mutations is located in the region where C-terminal part of the Negative Regulatory Region (HD-N) interacts with the juxtamembrane peptide (HD-C) by forming antiparalel beta sheets, or in a proximal alpha helix on the HD-N chain. Disruptions in this structure may loosen the interaction and lead to dissociation of the subunits and aberrant signalling.




