Function
ErbB2(also known as HER2 in human or Neu as an oncogene) is a tyrosine kinase belonging to epidermal growth factor receptor family. It is an orphan receptor, its function in the cell lies in heterodimerization with other members of its family – EGFR, ErbB3 and ErbB4 as a coreceptor. There it acts in transduction of signal by participating in transphosphorylation of kinases intracellular domain. It was found to be preferred interaction partner to other kinases from epidermal growth factor receptor family [1]. It also amplifies the signal and increases affinity for the ligands of its interaction partners [2]. From all its family members, it has the strongest catalytic activity. Effects of this kinase are mostly associated with promotion of proliferation, survival, and cell motility [3].
Structure
ERBB receptors contain an extracellular domain (ECD), a transmembrane domain (TMD), an intracellular region that consists of a juxtamembrane domain (JMD), a kinase domain (KD) and a carboxy terminal tail domain (CTD) [4]. The ECD is comprised of four subdomains (I-IV). In the absence of ligand, the ECD adopts an auto-inhibited tethered (closed) conformation that involves domain II and IV. Upon ligand binding between domains I and III, the dimerization arm in domain II is untethered, leading to receptor homo or heterodimerization, allosteric kinase activation, CTD phosphorylation and downstream signaling [5][6].
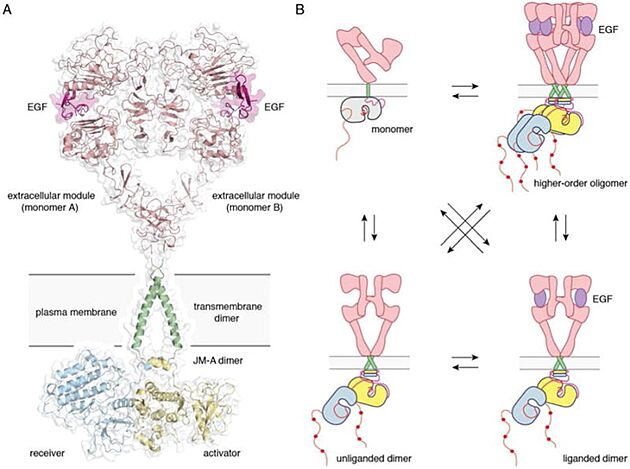
Full-length EGFR and its oligomerization states A) A proposed composite model of full-length EGFR based on the structures of individual modules (PDB ID 3NJP for the extracellular module, PDB ID 2M20 for the transmembrane - JM-A helices, and PDB ID 2GS6 for the kinase domains). B) Schematics for possible oligomerization states of EGFR in cells.
[7]HER2 is an atypical member of the ERBB family, as its ECD adopts an untethered conformation constitutively [8]. Unlike the other ERBB family members, HER2 does not have a ligand. HER2 preferentially heterodimerizes with ligand bound untethered (open) HER3 or EGFR to initiate cellular signaling, although HER2 homodimers capable of signaling have been reported in HER2 overexpressing cells [9][10][11]
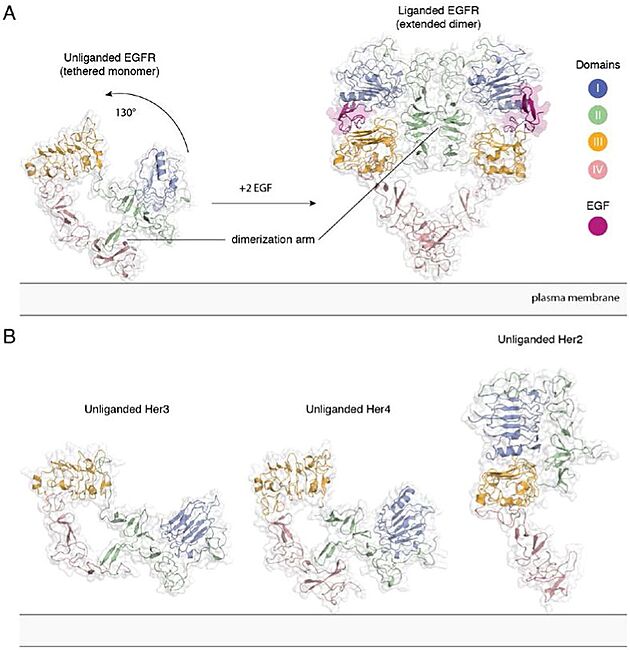
Extracellular module structures for the EGFR family members A) The conformational change induced by ligand binding. The tethered conformation of EGFR (left, PDB ID 1NQL, EGF bound at low pH was removed for clarity) rearranges to the extended conformation of EGFR (right, PDB ID 3NJP) upon ligand binding. B) Unliganded Her3 (PDB ID 1M6B) and Her4 (PDB ID 2AHX) can adopt a tethered conformation similar to EGFR, while Her2 (PDB ID 1N8H) is in an extended conformation, even in the absence of ligand.
[12]
Disease
Because of its properties, ErbB2 can play a role in cancer development. Its aberrant activity has been associated with breast cancers, in about 20 % of patients [13]. The cause is in majority ErbB2 gene amplification, its expression is in some cases increased up to 100-fold [14]. Though amplification is the major cause of ErbB2 mediated cancer, there have been found numerous mutations that contribute to cancer development, particularly promoting dimerization of kinase [15]. Recent large-scale sequencing efforts have identified oncogenic mutations in the ECD and KD [16][17][18]. Mutations in the TMD and JMD have also been reported, albeit at a low frequency [19][20][21][22][23]. Somatic mutations can follow after ErbB2 gene amplification, though they arise only in about 3 % of cases of breast cancer [24] . They are also found in other types of cancer, most frequently in bladder, cervical and ampullary cancer [25].
TMD/JMD mutations function by improving the active dimer interface or stabilizing an activating conformation.
TMD possesses an amphiphilic quality resulting from a stretch of polar residues along one face of the N-terminus of the TMD helix (S649, T652, S653, S656. The most frequent mutations identified in patient data set (V659E and G660D/R) extend this polar strip. This observation implies the polar character of these mutations amplifies intrinsic properties of the native TMD which may be an essential feature leading to the activating effect of these mutations on HER2. [26]
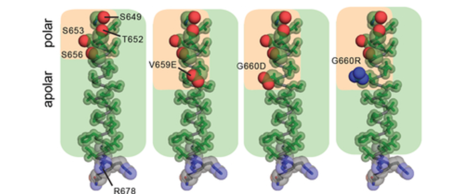
HER2 mutations in patient tumors. Amino acid composition of the TMD in WT HER2 (PDB ID: 2JWA) and in V659E, G660D, or G660R mutants, highlighting the relative arrangement of side chain atoms of polar (oxygen (red) and nitrogen (blue) atoms shown as spheres) and apolar (carbon atoms (green) shown as sticks)
[27]Proper positioning of the S656-xxx-G660 motif for productive TMD dimer formation is highly dependent on the orientation and geometry of the monomeric TMD helices, defined by basic residues near the interfacial regions between the cytoplasm and head group region of the bilayer [28][29][30]. Activating HER2 mutations such as R678Q might have a significant effect on the TMD geometry and dimerization. This was tested by performing all-atom 100 ns MD simulations for wild-type (WT) and the WT/R678Q HER2 TMD dimers in a phospholipid bilayer. The coordinates of the HER2 TM dimer in the putative activated conformation determined by NMR (PDB ID: 2JWA) were used as the starting positions in the simulations. The conformation of the WT HER2 TMD homodimer (WT/WT) remains stable over the course of the simulation. In the WT/R678Q TMD heterodimer, the S656-xxx-G660 motif remained engaged for the duration of the simulation, albeit through different interactions. However, the R678Q containing region of the C-termini separated by several angstroms compared to the WT homodimer (Figure 4). Despite these differences, in both WT/WT and WT/R678Q dimers, the conformations observed in the final state are consistent with a geometry proposed to support an activated, asymmetric configuration of the cytoplasmic kinase domains, and suggests that the enhanced activity of the mutant may be the result of its stabilizing effect on the specific heterodimeric configuration required for signaling.
A second possibility is that oncogenic mutations are able to stabilize an alternate activated dimeric TMD conformation. It is well known that polar interactions strongly support helix association both in conformations that cooperate with small Sm-xxx-Sm motif dimerization or in entirely unique geometries [31][32][33]. To see the effect of a polar mutation on HER2 TMD dimers the G660D mutant was simulated. MD simulations demonstrated that the introduction of the protonated aspartate disrupts the native dimeric configuration (Figure 4). In five independent 100 ns simulations, the TMD dimer configuration gradually drifted away from the starting configuration sampled for WT/WT HER2 and without achieving a common final state. On a 100 ns time scale it was not possible, however, to predict with certainty the final geometry of a HER2 TMD dimer in the presence of the G660D/R mutations, but these results suggest that polar mutations at position 660 alter the WT HER2 TMD geometry.[34]
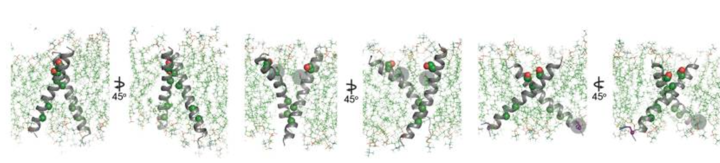
Conformational analysis of HER2 TMD mutants. Overview of the final state obtained at the end of a 100 ns simulation of the WT HER2 TMD dimer (left), a heterodimer between a WT TMD and an activating C-terminal R678Q TMD mutant (WT/R678Q) (middle) and a homodimer of the activating N-terminal G660D TMD mutant (G660D/G660D) (right).
[35]To understand the activating effect of the juxtamembrane latch mutant, Q709L, the interface between the juxtamembrane latch and the C-lobe of the activator kinase was analysed in a model of the HER2 homodimer constructed using the crystal structure of the EGFR/HER3 heterodimer (PDB ID: 4RIW) in which HER3 in the activator position was replaced with the HER2 kinase domain (PDB ID: 3PP0), and the juxtamembrane latch sequence of EGFR in the receiver position was replaced with that of HER2 (Figure 5). From this model it was observed that Q709 does not optimally participate in the dimer interface. Calculation of the HER2 C-lobe electrostatic surface reveals a hydrophobic pocket in the vicinity of the large polar side chain of Q709 likely suboptimal for hydrophobic packing between the C-lobe and the juxtamembrane latch. In contrast, the presence of a hydrophobic side chain such as leucine appears to be more optimal for favorable interactions with such a hydrophobic pocket, and is predicted to stabilize the dimerization interface essential for the allosteric activation of HER receptor kinase activity.[36]
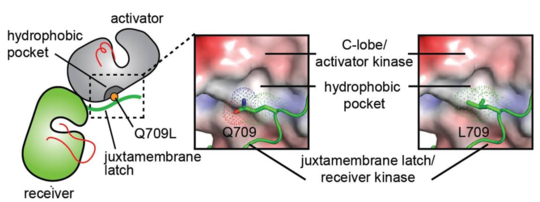
Conformational analysis of HER2 JMD mutants. Surface representation of the C-lobe of the HER2 kinase domain bound to a juxtamembrane latch binding region to depict interactions mediated by Q709 and L709. The interface was modeled by aligning structures of the HER2 kinase domain (PDB ID: 3PP0) on both activator and receiver kinases in the structure of the HER3/EGFR asymmetric dimer (PDB ID: 4RIW).
[37]





