References: the use of JSmol in Proteopedia [1] or to the article describing Jmol [2] to the rescue.
Biological Role
(VKOR) is a reducing enzyme composed of 4-helices that spans the endoplasmic reticulum as a transmembrane protein
[3]. Its enzymatic role is reducing (KO) to Vitamin K Hydroquinone (KH2)
[4] (Figure 1). The mechanism first occurs through the binding KO and using two cysteine residues to reduce KO into
Vitamin K. Then, a second pair of cysteine residues will reduce Vitamin K into the final product, KH2 (Figure 1). One of VKORs primary roles is to assist in blood coagulation through this KH2 regeneration mechanism.
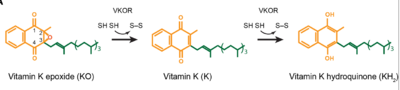
Figure 1. Mechanism of KO reduction into KH2.
With Vitamin K as a cofactor, the
γ-carboxylase enzyme will enact post-translational modification on KH2, oxidizing it back to KO
[5]. The oxidation of KH2 by γ-carboxylase is coupled with the carboxylation of a glutamate residue to form γ-carboxyglutamate. The coupling of this oxidation and carboxylation will activate several clotting factors in the coagulation cascade.
Author's Notes
Structural characterization of VKOR has been difficult due to its in vitro instability. Recently, a series of atomic structures have been determined utilizing anticoagulant stabilization and VKOR-like homologs[6]. Crystal structures of VKOR were captured with a bound substrate (KO) or vitamin K antagonist (VKA)[7]. VKA substrates utilized were anticoagulants, namely Warfarin, Brodifacoum, Phenindione, and Chlorophacinone. Second, VKOR-like homologs were utilized to aid in structure classification. Homologs refer to specific cysteine residues that have been mutated to serine to facilitate capturing a stable conformation state. Homologs were mainly isolated from human VKOR with some isolated from the pufferfish Takifugu rubripes. Homologs were also tagged at the N and C with a superfolder Green Fluorescent Protein(sfGFP)[8]. The sfGFP provides in vitro stability, a scaffold for crystallization, and facilitates in structure determination. Also, the sfGFP induces states of catalytic activity and potential inhibition for VKOR homologs. For the purpose of this report, all of the structures used have been processed to remove the sfGFP at the south end of VKOR as sfGFP served no purpose in function of the enzyme. This removal allowed for the residue numbering to be reassigned and more closely replicate the human VKOR.
Structural Highlights
VKOR has many key structural components that allow it to maintain proper functionality and catalytic abilities. The main part of the enzyme that contains the active site is a binding pocket where main catalytic activity occurs. The VKOR binding pocket provides specific substrate binding via highly conserved residues that recognize the target substrates. The pocket works in conjunction with the cap domain. The cap domain is a helical component of VKOR that facilitates conformational transitions from the to the once a substrate binds. Interactions between the cap domain, binding pocket, and the bound protein are critical to achieve full activation of Vitamin K. Another necessary part of the structure is the anchor. The anchor serves as a way to hold VKOR in the proper orientation within the cell membrane such that all enzymatic components are in the correct proximity for substrate binding and catalysis. Vital to the VKOR structure and these components are two disulfide bridges. The first appears slightly above the binding pocket between C132 and C135. The second occurs within the cap domain between C43 and C51. These cysteines are catalytic residues that also aid in the transition of VKOR from the open conformation to the closed conformation and the reduction of KO.
Cap Domain
Anchor
Active Site
Within the four transmembrane helices lies the . The binding pocket holds two active site residues, N80 and Y139, that interact with the substrate. N80 and Y139 are surrounded by a that provides specificity to the region. The hydrophilic residues hydrogen bond to the substrate, providing recognition and increasing specificity. The above the binding pocket provides stabilization when a substrate is bound. This bridge provides increased stability for the binding site as it interacts with and binds substrates or inhibitors. The hydrophilic residues provide when interacting with substrates for specificity and recognition. Upon binding, VKOR will transition into the closed conformation allowing the catalytic mechanism to commence.
Catalytic Mechanism of VKOR
Brief Overview
The overall mechanism works to convert Vitamin K epoxide to an activated form of Vitamin K hydroquinone, as noted in Figure 1. The substrate will bind VKOR at the binding pocket in the and induce the . Transition from open to closed conformation occurs with the oxidation of the C43-C51 disulfide bridge. Here, VKOR will utilize the second pair of , C132 and C135, to reduce KO into Vitamin K and Vitamin K into KH2. KH2 will be released from the binding fully activated and ready for use in the body. VKOR will reset, returning to the open conformation again, prepared for another substrate to bind.
Enzymatic Mechanism
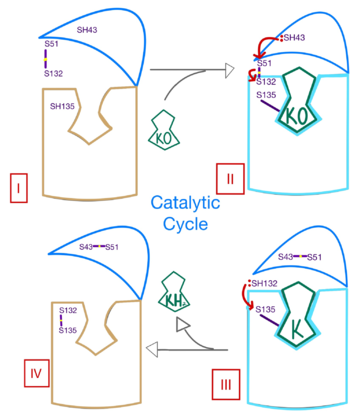
Figure 3. Mechanism of VKOR.
The catalytic mechanism of VKOR is highly regulated and use to activate Vitamin K necessary for blood coagulation. Figure 3 highlights these reactions that allow the substrate to be catalyzed to its active form through a series of 4 stages. The enzyme begins in in the open conformation with the cap domain open to allow substrate binding. Once a substrate binds, the cap domain transitions to the closed conformation, initiating when the C51-C132 disulfide bridge is attacked by reactive C43 located within the cap domain. This reaction forms a new disulfide bridge between C43 and C51 that pulls the cap domain over the binding pocket with the substrate bound to stabilize the closed conformation of VKOR. VKOR is now in stage II. Free cysteines are now available that provide strong stabilization of the closed conformation through interactions with the cap domain and the bound substrate. This puts the enzyme in , where a free C135 is purposed to interact with the substrate within the binding pocket to stabilize it during activation. The catalytic free C132 located between the cap domain and helical tunnel is very reactive and will attack this C135 to break that interaction with the substrate and release the activated Vitamin K product into the blood stream to promote coagulation. Two very stable disulfide bridges between C43-C41 and C132-C135 are now present and VKOR is unbound, so the enzyme is in its final, unreactive . VKOR must undergo conformational changes to return to Stage 1 and reactivate its catalytic cysteines so that another molecule of Vitamin K can bind and be activated.
Disease and Treatment
Afflictions
Since activated Vitamin K plays a crucial role in blood coagulation, defects in the function and enzymatic activity of VKOR may detrimentally effect on Vitamin K's ability to promote blood clotting. Mutations in VKOR also increase susceptibility to vascular diseases, such as a stroke [9]. Vitamin K is also important in maintaining bone health with inactivity of VKOR linked to decreased bone density and osteoporosis osteoporosis[10].
Inhibition
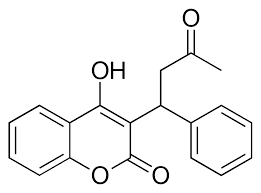
Figure 4. Structure of Warfarin.
The most common way to treat blood clotting is using the VKOR inhibitor, . Warfarin outcompetes KO[11], such that Vitamin K cannot be activated to promote coagulation in the blood. Warfarin will enter the binding pocket of VKOR, creating strong with the active site residues, N80 and Y139. Mutations of VKOR can lead to warfarin resistance which decreases its anticoagulation effects. Different mutations introduce varying degrees of resistance. These mutations are important to recognize as super-warfarin's can be overly effective in anticoagulation and become detrimental to blood flow.
Mutations
Mutations of the can occur within the binding pocket of VKOR. These mutations can be detrimental to the VKOR structure and function[12]. Two of the most common mutations occur to residues N80 and Y139 mutating them to . The change in polarity of these mutations from polar to nonpolar will cause a decrease in recognition and stabilization due to the inability to provide hydrogen bonds.



