This old version of Proteopedia is provided for student assignments while the new version is undergoing repairs. Content and edits done in this old version of Proteopedia after March 1, 2026 will eventually be lost when it is retired in about June of 2026.
Apply for new accounts at the new Proteopedia. Your logins will work in both the old and new versions.
User:Laura Carbone/Sandbox 1
From Proteopedia

Green fluorescent protein (GFP) is a bioluminescent polypeptide consisting of 238 residues isolated from the body of Aequorea victoria jellyfish.[1] GFP converts the blue chemiluminescent of aequorin in the jellyfish into green fluorescent light.[2] In remains unclear why these jellyfish use fluorescence, why green is better than blue, or why they produce a separate protein for green fluorescence as opposed to simply mutating the present aequorin to shift its wavelength,[3] but in the laboratory, GFP can be incorporated into a variety of biological systems in order to function as a marker protein. Since its discovery in 1962, GFP has become a significant contributor to the research of monitoring gene expression, localization, mobility, traffic, interactions between various membrane and cytoplasmic proteins, as well as many others.[4]
Contents |
History
Aequorea victoria was first discovered and investigated for its bioluminescence by Frank Johnson, who invited Osamu Shimomura to work with him in on a small island not far from British Columbia, where the jellyfish is abundant.[5] Found off the west coast of the United States between British Columbia and central California,[6] the jellyfish was considered a local phenomenon as it would drift in and out of the harbors.[5]
Shimomura was originally looking only to isolate the blue luminescent protein of Aequorea victoria, traditionally thought to be luciferase, but it would soon become apparent that the glow was in fact due to aequorin, a substance related, but slightly varying from luciferase.[4][5] However, the light emitted from aequorin still differed from the light emitted from the wild jellyfish. This quandary led to the discovery of the green fluorescent protein responsible for this disparity, but sufficient amounts of the protein could not be collected for study until 1979. The journey to discover the nature of GFP had begun.[5]
In the 1990’s, Douglas Prasher, Frank Predergast, and co-workers successfully cloned the gene that encoded for GFP. Martin Chalfie further pursued this line of work and was eventually able to express GFP in heterologous systems such as E. coli and C. elegans. Chalfie’s research provided the first evidence that GFP was unique as it did not require the presence of any exogenous substance or cofactor for fluorescence.[4] The lack for the need for a cofactor proved that the cloned GFP gene contained all the information necessary for posttranslational synthesis of the chromophore. [3]
Roger Tsien and co-workers were intrigued by the absence of a necessary cofactor and began to research the structure of GFP and how it relates to its fluorescence. They discovered that a helix within the beta barrel structure of GFP actually contained a fluorophore responsible for fluorescence. In researching its structure, they were able to develop GFP derivatives with improved fluorescence and photo-stability. Shimomura, Chalfie, and Tsien were each recognized for their work involving GFP with the Nobel Prize in 2008.[4] In the time since the work of these three researchers, GFP has been successfully expressed and utilized in bacteria, yeast, slime mold, plants, drosophila fruit flies, zebra-fish, and mammalian cells.[2] Below, mice have had GFP inserted into their genomes for studies in neurology.

Structure
Primary & Secondary Structure
| |||||||||
| 1ema, resolution 1.90Å () | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Non-Standard Residues: | , | ||||||||
| |||||||||
| |||||||||
| Resources: | FirstGlance, OCA, RCSB, PDBsum | ||||||||
| Coordinates: | save as pdb, mmCIF, xml | ||||||||
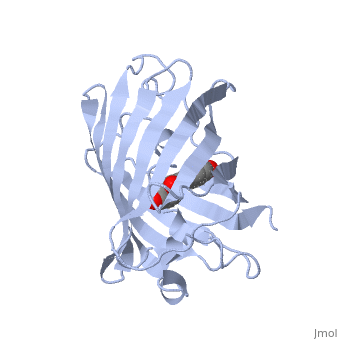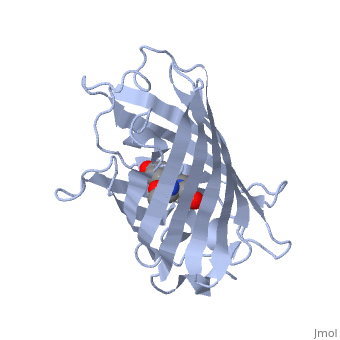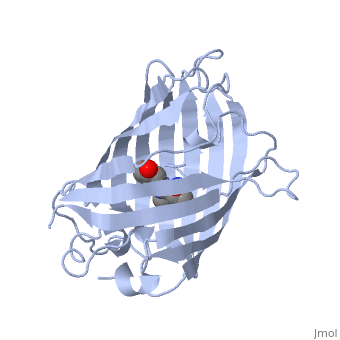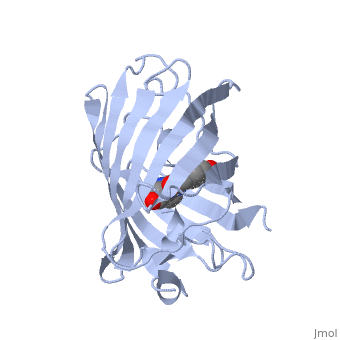

Green fluorescent protein () is a 21 kDa protein consisting of 238 residues strung together to form a of five α-helices and one eleven-stranded β-pleated sheet,[1] where each strand contains nine to thirteen residues each.[7] (To view the primary and secondary structure of GFP, go to [www.ebi.aci.uk].) These β-strands display an almost “seamless symmetry” in which only two of the strands vary in structural content.[8] This β-sheet conforms itself through regular hydrogen bonding into a β-barrel.[2] In GFP, the structure is so regular that of water molecules (red) can be seen following the structure of the barrel.[8] Together with the α-helices at either end of the molecule, a nearly perfect cylinder is produced, 42Å long and 24Å in diameter,[7] creating what is referred to as a “β-can” formation.[8] The short helical segments at either end of the cylinder form “caps” to further protect the interior of the β-barrel.[8] Overall stability is maintained by this β-can structure, helping to resist unfolding from heat and other denaturants.[2]
One can be found running through the central axis of the β-barrel,[4] roughly to the symmetry axis of the barrel.[7] This helix is extremely important as it contains the fluorophore responsible for fluorescence.[2][4] This α-helix in particular is highly stabilized by the many that are made with each strand of the barrel.[9]
The Chromophore
The of GFP is located at the center of the β-barrel with a wild-type excitation peak of 395 nm, and a minor peak at 475 nm (about three times less intense[3]) [2][10][7][8] with extinction coefficients of approximately 30,000 and 7,000 M-1 cm-1, respectively.[2][8] Interestingly, the Aequorea victoria jellyfish utilizes the smaller of the two excitation peaks as pure aequorin emits a light of 470 nm.[3] The relative amplitudes of these two excitation peaks can vary depending on environmental factors and previous illumination.[7] For example, continued excitation leads to a diminution of the 395 nm excitation peak with a reciprocal amplification of the 475 nm peak.[8] Regardless of absorption, the chromophore of GFP emits light of 508 nm.[2][10][7][8]
Three amino residues in the central α-helix constitute the fluorophore of GFP: Ser65Tyr66Gly67 (see left). Tsien et al. discovered that this tri-peptide sequence is post-translationally modified by internal cyclization and oxidation[4] to produce a structure.[2] Studies with E. coli proposed a sequential mechanism for the formation of the fluorophore that was initiated by a rapid cyclization between Ser65 and Gly67 to form an imidazolin-5-one intermediate.[2] This rapid cyclization is carried out via nucleophilic attack of the amino group from Gly67 on the carbonyl group of Ser65 to form a five-membered ring. The loss of water then forms the imidazolin-5-one intermediate.[10] Cyclization is succeeded by a much slower rate-limiting oxygenation of the Tyr66 hydroxybenzyl side chain by atmospheric oxygen (No fluorescence was seen in anaerobically grown E. coli.), resulting in the 4-(p-hydroxybenzylidene)-imidazolidin-5-one stucture.[2][10][8] The double bond that results from this series of reactions results in the linkage of the two π-systems of the rings, forming a larger conjugated system essential for fluorophore stability. [11]

The process is completely auto-catalytic such that there are no known co-factors or enzymatic components required.[2] Despite the stability of the final product, while the chromophore is forming, the environmental temperature cannot drop below 30°C or the yield of viable GFP will decrease substantially.[2][8] This, of course, is not an issue for the protein in nature as the jellyfish is unlikely to encounter waters of this degree in the Pacific Northwest.[3] Such a temperature sensitivity is only relevant during formation as the stability of the final product is maintained through a network of close contacts surrounding the fluorophore.[2] This, however, can and has been used in in pulse-chase experiments in which the GFP-expressing cells are exposed to varying temperatures in place of labeled vs. unlabeled trials.[3]
As the central α-helix is not located directly in the center of the β-barrel, cavities of differing area exist on either side of the chromophore. The larger cavity, consisting of about 135 Å,[7] does not open out to the bulk solvent, but rather houses .[7][12] Had this space not been occupied, it would be expected to considerably destabilize the protein as a whole. The hydrogen bonding created by the presence of the water molecules, however, helps to link the buried of Glu222 and Gln69 that would otherwise be actively polar.[7] Therefore, the water molecules are extremely important in establishing a hydrogen bonding network about the chromophor.[13]
|
The opposite side of the chromophore, however, is within close proximity of several aromatic and polar side chains. Several between the surrounding residues and the chromophore are present including: hydrogen bonds of His148, Thr203, and Ser205 with the phenolic hydroxyl of Tyr66; Arg96 and Gln94 with the carbonyl of the imidazolidinone ring; and hydrogen bonds of Glu222 with the side chain of Thr65. Additional hydrogen bonding in the area around the chromophore helps to stabilize Arg96 in the protonated form, which suggests the presence of a partial negative charge on the carbonyl oxygen of the imidazolidinone ring in the deprotonated fluorophore.[7] Arg96 and Gln94 in turn help to steady the imidazolidone.[2] Therefore, it is thought that Arg96 is essential for the formation of the fluorophore by catalyzing the initial ring closure.[7] Tyr145 provides a stabilizing with the benzyl ring of the chromophore.[7] (For more information about edge-face (CH/π) interactions, click here.) The stability provided by the internal polar interactions are further augmented by the surrounding β-barrel.
The β-barrel provides a highly constrained environment that protects the chromophore from the bulk solvent,[4] nearly creating the atmosphere of a vacuum.[13] This is most likely responsible for the small Stoke’s shift, or the small wavelength difference between excitation and emission.[7]
Findings show that fluorescence will not occur from a naked chromophore, but rather requires the protection of the β-can structure.[10] However, in crystallum GFP will exhibit a nearly identical fluorescence spectrum and lifetime when compared with aqueous GFP. These two elements point to a fluorescence that is not inherent to the isolated fluorophore,[2][8] but rather from the auto-catalytic cyclization of the polypeptide sequence Ser65Tyr66Gly67 and subsequent oxidation of Tyr66.[8] However, this sequence is found in many proteins - why does GFP fluoresce? According to Phillips (1997), fluorophore formation is due to the close proximity of the backbone atoms between Ser65. and Gly67 gained through a lack of sterical hindrance by the hydrogen atom side chain of glycine. In fact, no functional fluorescent proteins have been found in which any other amino acid other than glycine was found at position 67. Even so, there are still proteins that have this specific sequence, therefore, there must be another inherent property to GFP that is still left misunderstood.[8]
This quandary led Phillips to study the acid/base chemistry catalyzing the initial cyclization of the chromophore. He found that Arg96 actually acts as a by withdrawing electrons through hydrogen bonding with the carbonyl oxygen of Ser65 to activate the carbonyl carbon for nucleophilic attack by the amide nitrogen of Gly67. This mechanism was further supported by ab initio calculations, as well as database searches of similar compounds and protein sequences. Through acid/base chemistry, the chromophore is stabilized by resonance.[8]
Mutant Studies
|
Many mutant green fluorescent proteins have been developed in order to further understand the structure and mechanism of the fluorophore. The first mutagenesis studies simply of the amino acid sequence (. NOTE: The structure represented here is already truncated at the carbonyl terminus). Shortening the polypeptide by more than seven amino acids from either terminus lead to a total loss of fluorescence, as well as a complete failure to absorb light at the traditional wavelengths. This is most likely due to the structure of the protein. The last seven amino acid residues of the carboxyl terminus are roughly disordered, and thus do not interfere with the overall structure. After seven residues, however, the capping α-helix structure is disrupted, leading to an unstable or unformed chromophore. The is less understood, but the same principle still applies even though the β-barrel does not begin until residue ten or eleven.[2]
Point mutations have also been extensively studied in order to examine their effects on the chromophore. In general, most point mutations lead to a diminished excitation, especially in regions of the sequence adjacent to the fluorophore or those that interact with the fluorophore. An exception to this trend is the Ser65Thr66 mutant (normal Ser65Tyr66), which actually increases fluorescence intensity, although the reason is unclear.[2]
An interesting mutation discovered by Ormo et al. (1996) was the Thr65Tyr66Gly67 mutant, which produces an α-helical conformation in the chromophore opposed to the normal conformation, which is nearly perpendicular to the helical axis, due to its interaction with Arg96. This further supports the idea that Arg96 is an important factor in the structural arrangement required for cyclization, perhaps by promoting the attack of Gly67 on the carbonyl carbon of Thr65.[7]
In high protein concentrations, GFP has been found to dimerize under the influence of high ionic strength between the two monomers. In Aequorea victoria, the aequorin is able to bind to the (1gfl), but not the monomer. Therefore, dimerization is a very important structural feature in terms of its function, as it also assists the GFP to absorb energy at the excitation wavelength of aequorin even though GFP has only a “modest” extinction coefficient. As a result, dimers, and often even higher (1w7s), are predominant protein populations within the jellyfish.[10]
Use in the Laboratory
Green fluorescent protein has had great success as a marker protein in a variety of biological systems due to its inherent stability, which only adds to its many other desirable characteristics as a marker proteins.[4] GFP is rather resistant to denaturation,[2] sustaining structure and function up to 65°C, pH 11, 1% sodium dodecyl sulphate (SDS), or 6 M guanidinium chloride. GFP can also withstand the presence of most proteases for many hours.[10] Due to this stability, GFP could be applied to numerous other applications such as cell lineage tracing, gene expression reporting, or protein-protein interactions.[2]
While GFP can be incorporated into most prokaryotic systems, expression in eukaryotic systems may be limited to the cytoplasm and the nucleus, as GFP does not penetrate the nucleolus or vesicular organelles. However, highly specific intracellular localization can still be achieved in eukaryotes,[2] which can help to avoid the difficulties associated with adding extrinsic dyes.[12] However, this ability to generate fluorescence within live tissues in the absence of cofactors gives GFP the key for use in biological research. After an in-frame fusion to the protein of interest, the resulting chimeric protein can be expressed in a cellular environment to monitor function or activity[14].
Use of GFP as a Fusion Tag
One of the most common applications of GFP in research utilizes GFP as a fusion tag to a protein of interest in order to observe the activity of the protein. GFP is fused in frame with the gene encoding the protein of interest, resulting in a chimera that is both functional (hopefully) and fluorescent to be expressed in the organism. This technique has been used successfully in nearly every cell organelle, including the plasma membrane, nucleus, endoplasmic reticulum, Golgi apparatus, secretory vesicles, mitochondria, peroxisomes, vacuoles, and phagosomes. GFP is most commonly fused to either terminal end of the protein gene, although it may be possible to insert it onto a noncritical exterior loop or domain depending on the structure of the protein in question. For example, residues 2-233 of GFP were inserted between the last transmembrane segment and long cytoplasmic tail of a Shaker potassium channel in experiments done by Siegel and Isacoff (1997).[3]
Use of GFP as an Active Indicator
Because the β-can structure protects the chromophore from the surrounding environment, it is difficult to use wild-type GFP as an active indicator of changing conditions. However, environmental indicators have been created by combining various GFP mutants created from random and directed mutagenesis. It is also possible to incorporate phosphorylation sites into the GFP structure such that phosphorylation or dephosphorylation will induce major changes in fluorescence. For example, the Shaker fusion protein mentioned in the section above was the first genetically encoded optical sensor of membrane potential that induced at most a 5% decrease in fluorescence in phosphorylated conditions. However, the most general way to make biochemically sensitive GFPs is to exploit FRET as described in the following sections.[3]
Use of GFP in FRET
Förster resonance energy transfer (FRET) is a laboratory technique used to detect the proximity of two biological molecules by the fusion of fluorescent proteins with each of the proteins of interest. When the two fluorophores come within a certain proximity (typcially <100Å) in the proper orientation, the excited fluorophore (donor) emits energy that excites the second longer-wavelength fluorophore (acceptor) suc that it also fluoresces. Results can then be seen by the ratio of the donor and acceptor emission intensities. FRET is noninvasive and is therefore safe for using within live cells[14].
While the wild-type GFP protein has not been particularly useful as a sensor in FRET, several mutants of GFP have been manufactured to create proteins with distinct fluorescence spectra. FRET has been done between two GFP molecules or a single GFP molecule and a secondary fluorophore [14].
GFP Mutants Used In FRET
|
(Green links depict the chromophore of each GFP variant. The applet is currently showing the chromophore of .)
- GFP (eGFP): contains a mutation at Ser65 in which the residue is most commonly replaced by Thr, Ala, or Gly. Lacks the 395 nm excitation peak, but still emits light at 508 nm like wild-type GFP.
- (1bfp): blue-shifted GFP that has replaced Tyr66 with His (Y66H mutation).
- (1oxd): contains Y66H mutation with an emission spectra between BFP and eGFP.
- Sapphire: excitation spectra at 495 nm has been suppressed while the 395 nm spectra is retained; still emits light at 508 nm.
- (1yfp): red-shifted GFP that has replaced with an aromatic amino acid.
Förster Distances (nm) for Energy Transfer
| Acceptor (GFP variant or DsRed) | |||||
|---|---|---|---|---|---|
| Donor | Blue | Cyan | Green | Yellow | Red |
| Blue | 2.61 ± 0.05 | 3.77 ± 0.08 | 4.14 ± 0.08 | 3.82 ± 0.08 | 3.17 ± 0.06 |
| Cyan | - | 3.28 ± 0.07 | 4.82 ± 0.10 | 4.92 ± 0.10 | 4.17 ± 0.08 |
| Green | - | 1.93 ± 0.04 | 4.65 ± 0.09 | 5.64 ± 0.11 | 4.73 ± 0.09 |
| Yellow | - | 1.00 ± 0.02 | 3.25 ± 0.07 | 5.11 ± 0.10 | 4.94 ± 0.10 |
| Red | - | 1.40 ± 0.03 | 2.84 ± 0.06 | 3.14 ± 0.06 | 3.54 ± 0.07 |
- Data provided by [15]
Common Pairs of GFP Molecules Used in FRET
BFP-eGFP: The BFP-eGFP donor-acceptor pair is the most traditionally used pair of GFP molecules used in FRET, but BFP is only weakly fluorescent, creating a limitation for most applications aside from microscopy and flow cytometry.
CFP-YFP: This pair has been used more recently as an alternative to the BFP-eGFP pair because CFP is significantly brighter than BFP, allowing for more accurate ratiometric measurement of donor to acceptor emissions. However, filters must be used carefully while using CFP-YFP in FRET because of the bleeding of the CFP spectra into the YFP spectra. Therefore, proper filters must be set in order to distinguish the fluorescence from donor versus acceptor [14].
Related Structures and Topics
- 1ema Aequorea victoria Green Fluorescent Protein, monomer
- 1gfl Aequorea victoria Green Fluorescent Protein, dimer
- 1w7s Aequorea victoria Green Fluorescent Protein, wild-type tetramer
- 1emb Aequorea victoria Green Fluorescent Protein, Gln80 replaced with Arg
- 1b9c Aequorea victoria Green Fluorescent Protein Mutant F99s, M153t And V163a
- 1qxt Crystal structure of precyclized intermediate for the green fluorescent protein R96A variant (A)
- 1qy3 Crystal structure of precyclized intermediate for the green fluorescent protein R96A variant (B)
- 1qyo Anaerobic precylization intermediate crystal structure for S65 Y66 GFP variant
- 1qyq Crystal Structure of the cyclized S65 Y66 GFP variant
- 1jbz Crystal Structure analysis of a dual-wavelength emission green fluorescent protein variant at high pH
- 1jby Crystal Structure analysis of a dual-wavelength emission green fluorescent protein variant at low pH
- 1bfp Aequorea victoria Blue Variant of Green Fluorescent Protein
- 1yfp Aequorea victoria Yellow Variant of Green Fluorescent Protein
- 1huy Crystal structure of citrine, an improved yellow variant of green fluorescent protein
- 1oxd Aequorea victoria Cyan Variant of Green Fluorescent Protein
Reference for this Structure
Ormo M, Cubitt AB, Kallio K, Gross LA, Tsien RY, Remington SJ. 1996. Crystal structure of the Aequorea victoria green fluorescent protein. Science. 273(5280):1392-1395. DOI 10.1126/science.273.5280.1392.
References
- ↑ 1.0 1.1 [1], Protein Database (PDBsum): 1ema. European Bioinformatics (EBI); 2009.
- ↑ 2.00 2.01 2.02 2.03 2.04 2.05 2.06 2.07 2.08 2.09 2.10 2.11 2.12 2.13 2.14 2.15 2.16 2.17 2.18 2.19 2.20 [2], Yang F, Moss LG, Phillips GN Jr. 1996. The molecular structure of green fluorescent protein. Biotechnology. 14: 1246-1251. DOI 10.1038/nbt1096-1246.
- ↑ 3.0 3.1 3.2 3.3 3.4 3.5 3.6 3.7 Tsien, Roger Y. 1998. The Green Fluorescent Protein. Annual Review in Biochemistry. 67:509-544.
- ↑ 4.0 4.1 4.2 4.3 4.4 4.5 4.6 4.7 4.8 [3], Haldar S, Chattopadhyay A. 2009. The green journey. J Fluoresc. 19:1-2. DOI 10.1007/s10895-008-0455-6.
- ↑ 5.0 5.1 5.2 5.3 [4], Shimomura O. The discovery of green fluorescent protein. Nobel Prize Lecture; 2009.
- ↑ [5],Cowles D, Cowles J. Aequorea victoria. 2007. Walla Wall University.
- ↑ 7.00 7.01 7.02 7.03 7.04 7.05 7.06 7.07 7.08 7.09 7.10 7.11 7.12 7.13 Ormo M, Cubitt AB, Kallio K, Gross LA, Tsien RY, Remington SJ. 1996. Crystal structure of the Aequorea victoria green fluorescent protein. Science. 273(5280):1392-1395. DOI 10.1126/science.273.5280.1392.
- ↑ 8.00 8.01 8.02 8.03 8.04 8.05 8.06 8.07 8.08 8.09 8.10 8.11 8.12 8.13 Phillips GN Jr. 1997. Structure and dynamics of green fluorescent protein. Curr Opin Struct Biol. 7(6):821-827. DOI 10.1016/S0959-440X(97)80153-4.
- ↑ Andrews BT, Gosavi S, Finke JM, Onuchic JN, Jennings PA. 2008. The dual-basin landscape in GFP. Proceedings of the National Academy of Sciences. 105(34):12283-12288. DOI 10.1073/pnas.0804039105.
- ↑ 10.0 10.1 10.2 10.3 10.4 10.5 10.6 [6],Cubitt AB, Heim R, Adams SR, Boyd AE, Gross LA, Tsien R. 1995. Understanding, improving, and using green fluorescent protein. Trends in Biochemical Sciences. 20(11): 448-455. DOI 0.1016/S0968-0004(00)89099-4.
- ↑ Bublitz G, King BA, Boxer SG. 1998. Electronic structure of the chromophore in green fluorescent protein (GFP). Journal of the American Chemical Society. 120(36): 9370-9371. DOI 10.1021/ja98160e.
- ↑ 12.0 12.1 van Thor JJ, Sage, JT. 2006. Charge transfer in green fluorescent protein. Photochemical & Photobiological Sciences. 5:597-602. DOI 10.1039/b516525c.
- ↑ 13.0 13.1 Lammich L, Petersen MA, Nielsen, MB, Andersen LH. 2007. The gas-phase absorption spectrum of a neutral GFP model chromophore. Biophysical Journal. 92: 201-207. DOI 10.1529/biophysj.106.093674.
- ↑ 14.0 14.1 14.2 14.3 [7], Pollock, BA, and Heim, R. 1999. Using GFP in FRET-based applications. Trends in Cell Biology. 9 (2): 57-60. DOI 10.1016/SO962-8924(98)01434-2.
- ↑ [8], Patterson, GH, Piston, DW, and Barisas, BG. 2000. Förster Distances between Green Fluorescent Protein Pairs. Analytical Biochemistry. 284 (2): 438-440. DOI 10.1006/abio.2000.4708.
Additional Resources
- First Glance
- PDBsum: 1ema
- RCSB PDB 1ema
- OCA
- UniProt: P42212
- Scop: P42212
- CATH: 1emaA00
- Pfam: PF01353
- InterPro: IPR000786
- GFP featured at the Molecule of the Month series of tutorials by David Goodsell.
