Human Immunodeficiency Virus-1 Protease
Introduction
HIV -1 protease (HIV PR ) is a retroviral aspartyl protease that is derived from HIV-1, a lentivirus that is best characterized for its ability to lower host immunity by infecting CD4+ T lymphocytes, macrophages, and dendritic cells.[1] Aspartyl proteases are protease enzymes that utilize aspartate residue(s) for the catalysis of peptide substrates. Eukaryotic forms of these proteases include the , cathepsins and renins. While they have a two-domain structure, the retroviral aspartyl proteases are much smaller and homologous to a single domain of the eukaryotic aspartic proteases.
HIV-1 protease is essential for the life cycle of HIV. The protease takes newly synthesized polyproteins and cleaves them by means of a hydrolysis reaction into the smaller mature protein components of the HIV virion.[2] These proteins are used to form the virion capsid that encases the viral genome. Effectively inhibiting HIV-1 protease will result in the inability for HIV to propagate as it will no longer have the ability to form complete virion units to infect new cells. The HIV PR, together with single stranded RNA (ssRNA), reverse transcriptase, integrase, and other viral factors, is found inside the HIV-1 virion.[3]
[4]
Structure & Function
Structure of HIV-1 Protease
In the mid 1980's, the structure of HIV-1 Protease was hypothesized by Lawrence Pearl and William Taylor to consist of a single domain of the eukaryotic aspartic protease and function in the dimeric form.[5] As NMR was not yet in use at this time, x-ray crystallography was implored but required a large quantity of large crystals. This was problematic as heavy atom derivatives had to be used to overcome the phase problems with this new structure and a considerable amount of protein was needed. Eventually these problems were overcome and the first images of HIV-1 protease where announced by the Merck group, who were most successful at large-scale protein purification and crystallization.
Unlike most members of the aspartyl protease class, which generally exist as two domain monomers, HIV protease is a dimmer with two identical that are comprised of 99 amino acids. These two subunits come together to form a very narrow 7.46 angstrom diameter for the ligand to navigate; enclosing the active site in the middle. Instead of one flap (a long flexible β-hairpin loop from the N-terminal domain) closing over the active site like in pepsins, the homodimeric retroviral enzyme has two poorly ordered flaps.[6]The polyprotein is capable of making it to the active site not by squeezing through the narrow tunnel but rather by having the flaps . Each subunit donates the catalytic triad, Asp-Thr-Gly, to the highly conserved active site, which also closely resembles that of monomeric proteases like pepsin. The residues are Asp25, Thr26 and Gly27 on each of the subunits. The active site is in the retroviral aspartyl protease just as in the eukaryotic aspartyl proteases. As will be seen in the mechanism, the two will hydrolytically cleave the polyprotein that comes into the active site.
Mechanism
 Mechanism for HIV-1 Protease
Mechanism for HIV-1 Protease
The mechanism most commonly accepted for the aspartyl proteases is a general acid-base mechanism involving the coordination of a water molecule between the two highly conserved aspartate residues which leads to hydrolysis. One of the aspartate residues induces activation of the water molecule by removing one of the protons. This allows the nucleophilic oxygen to attack the carbonyl carbon of the peptide and form a tetrahedral oxyanion intermediate. Rearrangement leads to the protonation of the scissile amide and thus cleavage.
Applications & Research
Currently there is no cure for HIV. However after some extensive research, some drugs were to be designed in order to pause the HIV affects. This happens by the ability of the drugs to mimic the tetrahedral intermediates in the active site and prevent the polypeptide from binding the Asp residues. When the HIV infects an organism it tends to multiply within the body’s cells. The virus is then released to infect other cells. In this manner, the infection of HIV infects the newly made cells of the body. While the viruses are produced, proteins and enzymes used to manufacture the DNA in addition to other components of the virus are made. In this case, is that enzyme that is needed to bring the structural and enzymes of the virus together. Protease drugs are what could inhibit this virus.
- Design of a HIV-1 Protease inhibitor - Free-energy parameterization of enzyme-inhibitor binding.
Based on the crystal structure data and protein receptor ligand complexes studied, interatomic interactions that work on burying atoms and find the statistical preference for amino acid pairs. A free energy model of the receptor-ligand is formulated and helps in showing the interfacial interactions. The interaction strength of this model has a reliability of ±1.5 kcal/mol, which reveals the importance of atomic interaction to stabilize the receptor-ligand interface. The analysis of a binding motif of HIV-1 protease inhibitor complex shows the important contacts instead of the set of atoms. [7]
1) Saquinavir (Invirase) is known to be one of the first FDA approved protease inhibitor for HIV treatment. This usually occurs by HIV protease binding an active site , which will prevent polyproteins from also binding. Saquniavir's chemical structure has the ability to intermediate of the hydrolytic reaction to interact .
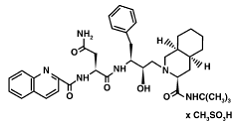
Saquinavir mesylate is a white to off-white, very fine powder with an aqueous solubility of 2.22 mg/mL at 25°C.
Knowing that, Saquinavir is an uncleavable ligand by studying its similar conformational changes in binding saquinavir or a polypeptide. [8] Two types of mutants can result as a way of depleting the effectiveness of the (Invirase) drug. These mutants are G48V and G48V/L90M. They cause 13.5 and 149 fold reductions, respectively. Based on some quantum mechanical and molecular mechanical analysis of the interaction of the saquinavir interaction; a disturbance of the saquinavir with the mutant with a flap residue 48 leads to a change in the hydrogen bonding. This mutation leads to the loss in the inhibitor/ enzyme binding.
2) Indinavir (Crixivan) this protease inhibitor is an oral medication that blocks the protease activity and leads to defect the viruses. Once these viruses are unable to reach the body’s cells, it will decrease in number.
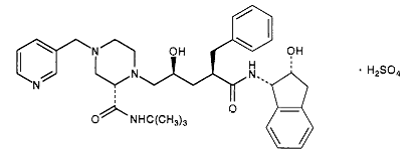
Ritonavir has a molecular weight of 711.88. It is very soluble in water and in methanol.
This inhibitor doesn’t have the ability to prevent the HIV transmission among individuals, nor does it cure AIDS or HIV infection. (Crixivan)is approved by the FDA in March 1995. [9]
3) Ritonavir (Norvir) , has the same effect of indinavir on HIV protease. It was approved by the FDA in June 1999. This drug consists of capsules or tablets 100 mg; Solution: 80 mg/ml. It is recommended that adults consume 600 mg twice a day and then increase by 100 mg in every 2 days.
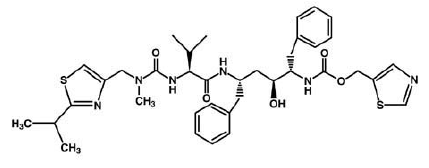
Ritonavir is soluble in isopropanol and practically insoluble in water.
(Norvir) can interact with many drugs leading those drugs to increase or decrease in effectiveness. It increases the concentration of Mycobutin and Viagra; however, decrease the concentration of Demerol.[10]
4) Nelfinavir (Viracept), Nelfinavir was approved by the FDA in March, 1997. (Viracept) has not been effectively evaluated in pregnant women. Viracept prevents T-cells that have been infected with HIV from producing new HIV. Usually given to patients through tablets. Each tablet contains inactive ingredients such as: calcium silicate, magnesium stearate, hypromellose, and triacetin.[11]
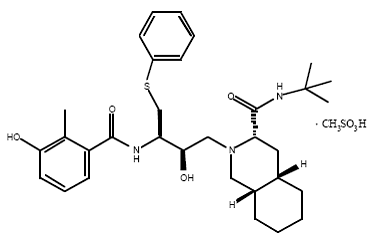
Nelfinavir is slightly soluble in water at pH ≤ 4 and completely soluble in methanol.
References
- ↑ Waki K, Durell SR, Soheilian F, Nagashima K, Butler SL, Freed EO. Structural and Functional Insights into the HIV-1 Maturation Inhibitor Binding Pocket. PLoS Pathog. 2012 Nov;8(11):e1002997. doi: 10.1371/journal.ppat.1002997. Epub, 2012 Nov 8. PMID:23144615 doi:10.1371/journal.ppat.1002997
- ↑ Nicholson LK, Yamazaki T, Torchia DA, Grzesiek S, Bax A, Stahl SJ, Kaufman JD, Wingfield PT, Lam PY, Jadhav PK, et al.. Flexibility and function in HIV-1 protease. Nat Struct Biol. 1995 Apr;2(4):274-80. PMID:7796263
- ↑ Winters MA, Lloyd RM Jr, Shafer RW, Kozal MJ, Miller MD, Holodniy M. Development of elvitegravir resistance and linkage of integrase inhibitor mutations with protease and reverse transcriptase resistance mutations. PLoS One. 2012;7(7):e40514. Epub 2012 Jul 18. PMID:22815755 doi:10.1371/journal.pone.0040514
- ↑ http://www.niaid.nih.gov
- ↑ Pearl LH, Taylor WR. A structural model for the retroviral proteases. Nature. 1987 Sep 24-30;329(6137):351-4. PMID:3306411 doi:http://dx.doi.org/10.1038/329351a0
- ↑ Miller M. The early years of retroviral protease crystal structures. Biopolymers. 2010;94(4):521-9. PMID:20593466 doi:10.1002/bip.21387
- ↑ Wallqvist A, Jernigan RL, Covell DG. A preference-based free-energy parameterization of enzyme-inhibitor binding. Applications to HIV-1-protease inhibitor design. Protein Sci. 1995 Sep;4(9):1881-903. PMID:8528086 doi:http://dx.doi.org/10.1002/pro.5560040923
- ↑ Mahajan SD, Roy I, Xu G, Yong KT, Ding H, Aalinkeel R, Reynolds J, Sykes D, Nair BB, Lin EY, Prasad PN, Schwartz SA. Enhancing the delivery of anti retroviral drug "Saquinavir" across the blood brain barrier using nanoparticles. Curr HIV Res. 2010 Jul;8(5):396-404. PMID:20426757
- ↑ . Indinavir (Crixivan). Treat Rev. 1996 Apr;(no 21):7. PMID:11363398
- ↑ . Ritonavir (Norvir). Res Initiat Treat Action. 2000 Mar;6(1):26-7. PMID:11708183
- ↑ Baker R, Bowers M. Nelfinavir (Viracept). BETA. 1997 Mar:5. PMID:11364528







