Highlighted Proteins of Lyme Disease
From Proteopedia
| Line 19: | Line 19: | ||
Researchers are attempting to take advantage of the upregulation of OspC on <i>B. burgdorferi</i>'s outer surface while the bacteria is in the host to develop an OspC-based vaccine. However, development of an OspC-based vaccination has been presented with difficulties due to its highly variable nature. <ref name='protein'>PMID:17921702</ref> | Researchers are attempting to take advantage of the upregulation of OspC on <i>B. burgdorferi</i>'s outer surface while the bacteria is in the host to develop an OspC-based vaccine. However, development of an OspC-based vaccination has been presented with difficulties due to its highly variable nature. <ref name='protein'>PMID:17921702</ref> | ||
| - | + | ||
<h3>Structure of OspC</h3> | <h3>Structure of OspC</h3> | ||
---- | ---- | ||
| + | {{STRUCTURE_1ggq| PDB=1ggq | SCENE=Studio:G4SecL04/Dimer_with_mg/1 }} | ||
The OspC three-dimensional model presented to the right is the <i>B. burgdorferi</i> B31 strain (residues 38-201) - also known as the oMG A strain. This is one of the four invasive oMGs that are responsible for systematic Lyme disease in mammalian hosts. In crystal structure, OspC exists as a dimer that coordinates a divalent ion, modeled here as magnesium. Each OspC subunit is predominantly helical, consisting of five parallel | The OspC three-dimensional model presented to the right is the <i>B. burgdorferi</i> B31 strain (residues 38-201) - also known as the oMG A strain. This is one of the four invasive oMGs that are responsible for systematic Lyme disease in mammalian hosts. In crystal structure, OspC exists as a dimer that coordinates a divalent ion, modeled here as magnesium. Each OspC subunit is predominantly helical, consisting of five parallel | ||
<scene name='Studio:G4SecL04/Helix_blue_in_ribbon/1'> α-helices </scene> | <scene name='Studio:G4SecL04/Helix_blue_in_ribbon/1'> α-helices </scene> | ||
Revision as of 11:40, 12 March 2014

Lyme disease is caused by three species of bacteria belonging to the genus Borrelia.[1][2] Borrelia burgdorferi, an obligate parasite, is the most common cause of the disease in the United States and is transmitted via hard-bodied ticks of the Ixodidae family, commonly known as the blacklegged or deer ticks. Borrelia spirochetes are motile, helical bacteria whose cell membranes have many exposed, surface lipoproteins that are involved in both the pathogenesis and life cycle of the parasite. Two predominant groups of the surface lipoproteins present are classified as outer surface proteins (Osps), which have been characterized as Osps A through F, and the variable major protein-like sequence expressed (VlsE). Both of these groups of outer surface proteins play important roles in both the pathogenesis and life cycle of Borrelia as well as roles in eliciting an immune response of the host organism (Figure 1).[3]
In a introductory biology course at Stony Brook University, undergraduates are modeling and exploring B. burgdorferi outer surface proteins and their respective antibodies in which the host organism produces. This Proteopedia page is the product of their efforts, with a focus on highlighted proteins from the following categories: OspC, OspB and the antibodies to OspB, OspA and the antibodies to OspA, and VlsE.
The goal of this Proteopedia page is to describe Lyme disease from a structural biology perspective in order to answer key questions regarding the relationship between the structure and function of B. burgdorferi proteins. What do B. burgdorferi outer surface proteins look like? How does the structure/function of these proteins relate to the infection cycle of B. burgdorferi? What are the structural targets of the human immune system and how have these targets evolved? What are the ideal structural targets for a vaccine to protect against Lyme disease?
Contents |
OspC and Lyme Disease

OspC, a highly variable B. burgdorferi outer surface protein, plays a pivotal role in the transmission of B. burgdorferi from the tick vector to a mammalian host. This protein is upregulated when the tick feeds, allowing for it to adhere to the tick saliva, move to the tick's mouth, and migrate into the mammalian host.[4] The upregulation of OspC is accompanied by a downregulation of two other outer surface proteins, OspA and OspB, which is thought to be induced by changes in environmental temperature and pH (Figure 2). [5]
Strains of B. burgdorferi are classified according to the sequence of the OspC locus into 19 outer surface major groups (oMGs), denoted by type A through S, of which only four are invasive (disease causing).[6] Polymorphisms of OspC and the abundance of invasive strains in a population of B. burgdorferi are driven by ecological factors, such as the mammalian host community composition, and are determinants of human Lyme disease risk[7].
Researchers are attempting to take advantage of the upregulation of OspC on B. burgdorferi's outer surface while the bacteria is in the host to develop an OspC-based vaccine. However, development of an OspC-based vaccination has been presented with difficulties due to its highly variable nature. [8]
Structure of OspC
| |||||||||
| 1ggq, resolution 2.51Å () | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ligands: | |||||||||
| Related: | 1f1m, 1osp, 1fj1 | ||||||||
| |||||||||
| |||||||||
| |||||||||
| Resources: | FirstGlance, OCA, RCSB, PDBsum | ||||||||
| Coordinates: | save as pdb, mmCIF, xml | ||||||||
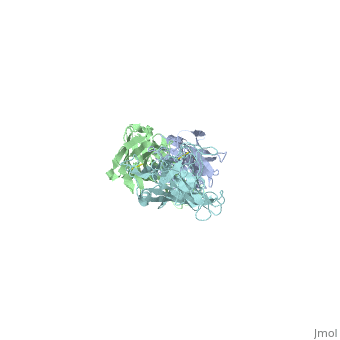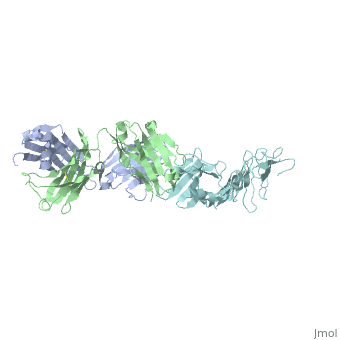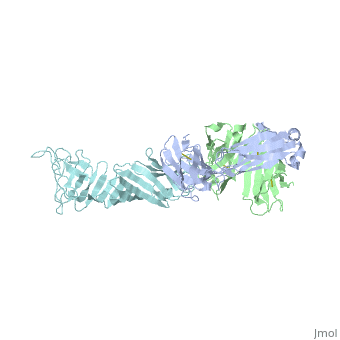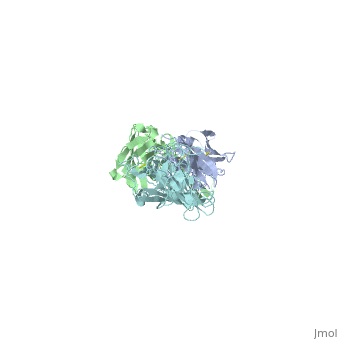

The OspC three-dimensional model presented to the right is the B. burgdorferi B31 strain (residues 38-201) - also known as the oMG A strain. This is one of the four invasive oMGs that are responsible for systematic Lyme disease in mammalian hosts. In crystal structure, OspC exists as a dimer that coordinates a divalent ion, modeled here as magnesium. Each OspC subunit is predominantly helical, consisting of five parallel , two short, antiparallel and six . The N and C termini at the membrane proximal end of two long α-helices, (residues 38-76) and (residues 170-201), are in close proximity to each other. At the membrane distal end, there are three remaining alpha helices, (residues 95-112), (residues 121-145), and a the final, short (residues 152-159). At the end of the membrane surface, the connection between helices α1 and α2 forms two short, anti-parallel, β-strands, (residues 79-80),and (residues 88-89).
While most of the OspC locus is highly variable, the sequence alignment of all oMGs reveals that the surface-exposed residues towards the membrane-proximal end of two of the helices, α1 and α5, are highly and have a positively charged surface. Other than those regions of the α1 and α5 helices, the surface-exposed residues on the remaining regions of the OspC molecule are variable.[9]

OspC Structure and Antigenicity
At the membrane-distal region, the six loop regions, including two β-strands, illustrates the of OspC with the presence of variable surface-exposed residues. [10] Among these variable regions, the outer surface-exposed residues connecting the helices α1 and α2, forming the loops, (residues 74-78), (residues 81-87), (residues 90-93), two short beta strands, β1 and β2, and (residues 146-150) are more variable than those present in the loops, (residues 115-119)and (residues 161-169). The surface potential of the red region that projects away from the membrane is negatively charged and primarily involved in the protein-protein or protein-ligand interactions.[9] Only four types of invasive oMG strains (A, B, I and K), whose surface potential in the red region is highly negative relative to non-invasive strains, play a major role in pathogenesis of human Lyme disease. [6]The residue, located in the red region at the distal, membrane end, is unique in that replacing this residue with other residues, with the exception of Lys82 and Gln82, which are only present in four invasive oMG strains, enhances the possibility of turning invasive strains into non-invasive strains. Thus, the stronger the negative electrostatic potential in the red region, the higher the chance OspC will to bind with positively charged host ligands; therefore, the altering of an amino acid residue at the 82nd position in the red region determines OspC polymorphism and demonstrates how this is connected to virulence and invasiveness.[6]
Lyme Disease and Ecology
![Figure 3: A Diagram of the Life Cycle of the Blacklegged Tick.[[1]]](/wiki/index.php/Image:Life_cycle_of_tick.png)
Ticks are born with the absence of the B. burgdorferi parasites and acquire them while feeding on the blood of natural, reservoir hosts, such as mice, squirrels, shrews, and other small vertebrates (Figure 3). After growth and development, the infected nymphal and adult ticks can transmit B. burgdorferi to incidental vertebrates, including humans. The ecological interaction between the competence of reservoir hosts and the ticks is an underlying measure of human Lyme disease risk.[11] The occurrence of Lyme disease is dependent upon the abundance of ticks that are infected with B. burgdorferi in natural ecosystems, and the number of reported cases of Lyme disease continues to increase annually in highly focused geographic locations of the United States (Figure 4).
Using Ecological Models to Predict Lyme Disease Risk
![Figure 4: Illustrated Prevalence of Lyme Disease in the United States (Generated by the CDC).[[2]]](/wiki/index.php/Image:Lyme_Disease_Risk_Map.gif)
Researchers have developed conceptual and mathematical models to characterize the ecological interactions between the community of vertebrate hosts and distribution frequency of invasive oMGs and predict the distribution of Lyme disease. In one effective model, the principal reservoir host used to calculate the risk of Lyme disease in northeastern and central United States is of the white-footed mouse (Peromyscus leucopus). The white-footed mouse has both a high frequency distribution in all four human infectious oMGs and high transmission probabilities of oMGs A, B, I and K.[7] It has been found that decreasing the abundance of mammalian hosts with high transmission probabilities, such as the white-footed mouse, and increasing other mammals with lower transmission probabilities drastically decreases the human Lyme disease risk. Many studies have found support for this "dilution-effect model," indicating that maintaining a high diversity of the vertebrate host community may be helpful in decreasing the incidence of Lyme disease. This model strongly demonstrates the relationship between species diversity in the community of hosts and the risk of human exposure to Lyme disease, and the ecological driving forces described in the model are useful tools in predicting the prevalence and risk of human Lyme disease.
Ecological factors responsible for human Lyme disease risk
The following are some factors used as parameters in ecological Lyme disease models:
- Vertebrate Community Composition[12]: Both the abundance and diversity of the mammalian hosts living in the community strongly affects the proportion of infected nymphal ticks that can cause human Lyme disease.
- Distribution frequency of particular oMGs[7]: As only four types of oMGs (A, B, I and K) are responsible for systemic human Lyme disease, the host-seeking nymphs that have a high distribution frequency of the four invasive oMGs is one of the standard measures of human Lyme disease risk.
- Transmission Probability[7]: The transmission probability of each oMG between individual species differs. The higher the transmission probability of a particular oMG from a vertebrate host, the higher the chances are that the ticks will carry that particular oMG after receiving a blood meal from their hosts. Thus, it is one of the parameters that contributes to the prevalence of human Lyme disease.
OspC-based vaccine
Researchers are currently developing an OspC-based vaccine against human Lyme disease, but because of the variability of OspC, the recombinant OspC vaccine, targeting the antigenic site of one B. burgdorferi strain, may be ineffective for other strains. Therefore, the development of a vaccine that recognizes the antigenic determinant on the variable regions of multiple OspC strains is required in order to effectively activate the human immune response. Based on the mapping of epitope-containing regions from oMGs A, B, K and D, the experiment-based tetravalent chimeric vaccine is being developed to trigger an anti-ABKD response. [13] Taking advantage of the tetravalent ABKD construct, an octavalent chimeric vaccine, also known as OspC-A8.1, recognizing additional epitopes of oMGs C, E, N and K has been tested in mice. [8]
OspB and Lyme Disease
OspB and OspA comprise the most common proteins found on the surface of B. burgdorferi while residing in the tick vector, during late-stage Lyme disease, and also during culture conditions.[14] OspB has been found to play a vital role in the adherence of B. bugdorferi to the tick midgut whereas OspB-deficient B. burgdorferi are shown to bind poorly to tick gut extracts. While the tick remains unfed, the expression of both OspB and OspA is upregulated to promote binding to the tick’s gut. However, during transmission from the tick to a vertebrate host, OspB is downregulated while other proteins, such as OspC, are upregulated [15]. OspB has shown significant variability in amino acid sequence and antigen reactivity in comparison to OspA, which is known to be largely invariant [16].

A factor contributing to the severity of Lyme disease is the ability of B. burgdorferi to evade the host immune system. B. burgdorferi is an extracellular pathogen, which is targeted by the humoral immune system of the host; the complement system and antibodies produced by B-lymphocyte-derived plasma cells work together in order to fight off the infection. The complement system consists of three pathways: the classical pathway, the alternative pathway, and the lectin pathway. Although each pathway differs in the process of initiation, each pathway results in the amplification of an immune response and the formation of the membrane attack complex (MAC), which ultimately leads to the pathogen's demise. In the lectin pathway and the alternative complement pathway, complement is recruited to the pathogen surface directly, but in the classical complement pathway, recruitment is dependent on the existence of an antibody-antigen immune complex; therefore, the traditional view of antibodies functioning in a bactericidal capacity has required complement recruitment and MAC formation following antibody binding to pathogens. Generally, antibodies are thought to be non-bactericidal in the absence of complement.
B. burgdorferi has developed resistance to a complement-dependent immune response by the evasion of the classical complement pathway [17][18] and the alternative complement pathway by binding complement components C4b and CS, respectively.[19] However, OspB is an important target of antibodies that can kill the bacteria without the help of the complement system.[3] These antibodies are termed complement-independent bactericidal antibodies. One important complement independent antibody whose bactericidal role has been well researched is CB2.[20] CB2 has been shown to kill bacteria in vitro in the absence of complement [21]. Furthermore, it has been shown that a point mutation in OspB renders the epitope unrecognizable by CB2, preventing CB2 from binding OspB and suggesting a possible mechanism for the evolution of the bacteria to evade this aspect of the immune system[22].
Another complement independent antibody is H6831, an IgG antibody that targets the C-terminal of OspB (Figure 6). Studies on both CB2 and H6831 have been conducted using the Fab (Fragment antigen binding) portion of these antibodies. A Fab consists of a heavy chain and light chain, each chain containing both a variable region as well as a constant region (Figure 5). The complementarity-determining regions (CDRs) are located at the N-terminal end of the variable region of the heavy and light chains of the Fab and form unique tertiary and quaternary protein structures that determine the antigen binding specificity. Binding of this region of the Fab to OspB of B. burgdorferi leads to the lysis of the bacteria, ruling out simple agglutination (clumping) of the bacteria as the cause of the bactericidal effect.[3]
Structure of the OspB-H6831 Complex
| |||||||
: ·· ·· ·· |
The consists of two components: outer surface protein B () and the , which is subdivided into the and the . Most hydrogen bonds and electrostatic interactions that are responsible for the binding of H6831 to OspB are between the at the C-terminal of OspB and some that include tyrosine, tryptophan, glutamate, and histidine.[16]
The majority of hydrogen bonds and electrostatic interactions are between (residues 250-254) and the Fab heavy chain. in loop 2 of OspB has a necessary and major role due to its central position in the exposed loops. A mutation at its position abrogates the binding interaction and causes the resistance of the bacteria to the bactericidal effect of the Fab. Lys 253 interacts with the two aromatic residues on the Fab heavy chain, tyrosine and tryptophan. It also makes hydrogen bonds with the glutamate 50 in the heavy chain of the Fab and forms an ionic bond. Carbonyl in of the OspB interacts with in the Fab heavy chain. of OspB interacts with Fab light chain.[16]
Structural changes to OspB in the complexed form
The binding of H6831 to OspB leads to some conformational changes in OspB compared to its . Crystallography has shown that the most significant difference is the loss of the .[16] The loss of these β sheets may be due to conformational change as a result of the binding or a disorder that could have occurred during a crystallization of the complex. Both small positional shifts near the Fab binding site and a few larger structural changes away from the binding site were observed. The largest shifts (7– 8 Å) correspond to the repositioning of a loop opposite the Fab-binding site . In the free OspB structure, all regions that exhibit shifts are adjacent to the central sheet; in the OspB-H6831 complex they all shift toward, and slightly overlap the position of the missing sheet.
Potential Mechanism of Lysis
The Fab binding destabilizes the outer membrane (OM) of B. burdorferi, with subsequent formation of spheroplasts. It has been observed that the bactericidal action, but not the binding, requires the presence of divalent cations (Mg2+ and Ca2+), and Fab is unable to clear bacteria in the absence of these cations.[23] It is speculated that OspB-Cb2 (a Fab similar to H6831) complexes could lead to the lysis of the cell by creating physical openings in the OM, allowing for rapid infusion of electrolytes and increasing the osmolarity of the periplasm.[24]
| |||||||||||


Proteopedia Page Contributors and Editors (what is this?)
Niamh O'Hara, Jonathan Manit Wyrick, Jaime Prilusky, Marvin O'Neal, Michal Harel
