Introduction
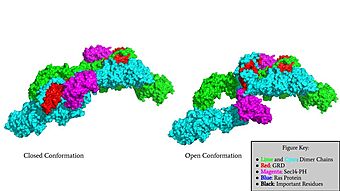
Surface Rendering of Neurofibromin in its Open and Closed Conformation
Neurofibromin is a protein that is coded for by the NF1 Gene which is located on chromosome 17. It functions as a tumor suppressor gene through its association with the protein Ras. The molecular structure of Neurofibromin has been determined by Cryo-Electron Microscopy. The structure of neurofibromin isoform 2 by cryo-electron microscopy revealed different functional states for the Neurofibromin protein.[1] Mutations in Neurofibromin is associated with diseases such as Plexiform Neurofibromas.
Structure
Neurofibromin is a made up of two identical chains. There are two conformations that classify neurofibromin known as its open and closed conformations. These conformations allow neurofibromin to associate with the protein Ras and perform its function of Ras regulation. The transformation between the overall closed conformation and open conformation of neurofibromin signifies a transition between an active neurofibromin protein and an inactive neurofibromin protein. There are two important domains involved in the transition between the open and closed conformations, the domain and the domain. Although neurofibromin is a homodimer with two identical protomers, only one protomer needs to have its GRD and Sec14-PH domains rotated in the open conformation in order for it to be able to perform its function.
Closed Conformation
The first conformation of Neurofibromin is known as the , which is representative of an inactive Neurofibromin protein. In the closed conformation, both sets of the GRD and Sec14-PH domains are rotated in a way that they are inaccessible and inactive due to a consisting of residues cysteine 1032, histidine 1558, and histidine 1576 that form a transition metal-binding site with zinc. The close proximity of the C1032, H1558, and H1576 residues that form the keep the GRD domain packed tightly on top of the Neurofibromin core. This tight compaction leads to steric inhibition when Neurofibromin tries to perform its function and associate with Ras. The association between Ras and Neurofibromin is supposed to occur via an located at Residue 1276. However, the steric hindrance from the Neurofibromin core in the closed conformation inhibits this association. Therefore, when Neurofibromin is in the closed conformation, there is no association with Ras and cell growth and proliferation is able to occur.
Open Conformation
The other conformation that characterizes Neurofibromin is the . In the open conformation of Neurofibromin, the protein is considered active and is participating in its function of Ras regulation. This occurs because the transition metal-binding site with zinc no longer is able to form due to an increase in distance between the C1032, H1558 and H1576 residues that form the . One protomer in Neurofibromin has its GRD and Sec14-PH domains oriented in way that is almost reversed in position compared to the closed conformation. Due to this rotation, C1032 is now located too far away, approximately 30 Angstroms, from H1558 and H1576 which results in the loss of the metal-binding site. The lack of the transition metal-binding site allows the GRD to orient itself in such a way that it can associate with . The reason that Neurofibromin is only able to associate with Ras in the open conformation is due to one critical residue, the located at position 1276 in Neurofibromin. When Neurofibromin is in the open conformation, R1276 is able to because there is no steric hindrance from the Neurofibromin core.
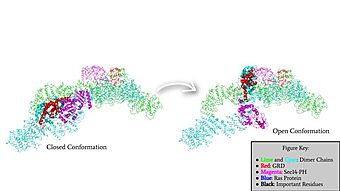
Rotation of the GRD and Sec14-PH domains from the closed conformation of neurofibromin to the open conformation of neurofibromin to allow Ras binding. The GRD rotates -130° and the Sec14-PH domain rotates -90°
Function
Neurofibromin functions as a tumor suppressor protein.[2] Its job is to prevent cell growth by turning off another protein known as Ras which in its active state, stimulates cell growth and division. Ras is a GTPase membrane protein that can only interact with Neurofibromin, a cytoplasmic protein, in the open conformation. As Neurofibromin is a cytoplasmic protein, it is brought to the membrane to associate with Ras via another protein known as SPRED1. Neurofibromin can interact with SPRED1 in both the open and closed conformations however, it can only associate with Ras when it is in its open conformation. The interaction between Neurofibromin and Ras occurs via an arginine finger (R1276) present in the GRD of Neurofibromin which is critical for Ras binding. R1276 is only accessible for binding when the GRD and Sec14-PH domains of Neurofibromin are rotated into the open conformation and there is no steric hindrance from the surrounding dimer chains. When R1276 is able to associate with Ras, Neurofibromin downregulates the Ras signaling pathway by hydrolyzing the GTP associated with Ras to GDP, effectively making it inactive and inhibiting cell growth and division.
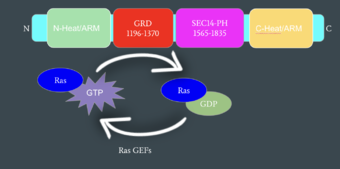
Mechanism of Ras Regulation by Neurofibromin
Disease and Medical Relevance
Currently, there are over 1485 mutations of Neurofibromin that have been identified. Mutations in Neurofibromin can lead to life-threatening illnesses or conditions such as Neurofibromatosis Type I due to the inability of Neurofibromin to interact with Ras.[3] The role of Neurofibromin is to inhibit cellular proliferation via its guanine triphosphatase-activating protein (GAP) activity, therefore when there are mutations to the NF1 gene that prevents the interaction of Neufibromin with Ras, there is nothing stopping Ras from promoting cell growth. Uncontrolled cell growth can lead to tumors and a higher risk of cancer. Neurofibromatosis Type 1, the most well-known disease resulting from mutations in Neurofibromin, is characterized by cognitive impairment, soft, non-cancerous tumors on or under the skin known as neurofibromas, birthmarks, clusters of freckles in unusual places, and problems with the bones, eyes and nervous system.
Neurofibromin is an essential protein and is involved mainly in the differentiation of neural crest derived cells, mesenchymal cells, neural cells, melanocytes, and bone cells. As Neurofibromin is essential for embryonic development, mutations to the NF1 gene can result in psychological retardation resulting from Type I neurofibromatosis. Most of the 1485 mutations identified lead to a synthesis of truncated, non-functional protein and are a result of point mutations. Type I Neurofibromatosis is inherited in autosomal dominant manner but about 50% of cases de novo ones. [4]
Student Contributors
- Hannah Luchinski
- Sophie Mullinix



