Introduction
Vitamin K epoxide reductase (VKOR) is the enzyme responsible for regenerating vitamin K from vitamin K epoxide to support blood coagulation.
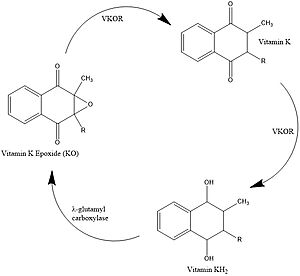
Figure 1. Vitamin K Cycle
Vitamin K Cycle
Vitamin K is essential for blood clotting in the body. The fully reduced form, KH2, allows the gamma carboxylation of blood clotting cofactors and is turned into the epoxide form in the process. Vitamin K epoxide reductase, abbreviated VKOR, turns the epoxide back to the fully reduced form so the reduced form can be used again. This transformation happens in two steps including converting the epoxide to the partially oxidized Vitamin K quinone then converting the quinone to the fully reduced hydroquinone (KH2). [1]
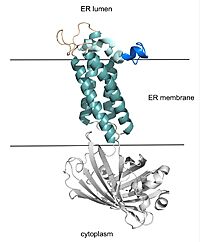
Figure 2. VKOR with Barrel Domain
Structural Overview
VKOR consists of four embedded in the endoplasmic reticulum membrane. The files on the RCSB Protein Data Bank include a barrel domain that is not pertinent to the function of VKOR. The images presented here have been edited to remove the barrel domain and renumbered to correspond with the article by Liu. [2]. The original image with the barrel domain in context is shown in Figure 2. Helices one and two are connected by the region which contains two of the active cysteines, C43 and C51. VKOR also has a covering the active site, made up of an , , and .
Active Site
VKOR uses two catalytic amino acids, tyrosine 139 and asparagine 80, to stabilize binding to all forms of and , such as Warfarin, in the binding pocket. Tyr139 and Asn80 hydrogen bond to carbonyl groups on both structures and stabilizes them within the binding pocket [2].
Other than the two previously mentioned hydrogen bonds (Tyr139 and Asn80), and are bound via hydrophobic interactions within the binding pocket of VKOR. Hydrophobic residues of VKOR such as Phe80, Phe87, and Tyr88, form a hydrophobic tunnel within the binding pocket [2].
Catalytic Cycle
Catalytic Cysteines
The catalytic cycle of VKOR includes transitions from open to closed conformations by means of disulfide bridge-induced conformational changes (Figure 3). Open conformations of VKOR exist when there is no ligand within the binding pocket. Closed conformations exist when some substrate exists within the binding pocket of VKOR. The substituent cysteines (Fig 3, step I) act as reducing agents for the substrate, which can be either Vitamin K epoxide (KO) or partially reduced Vitamin K.
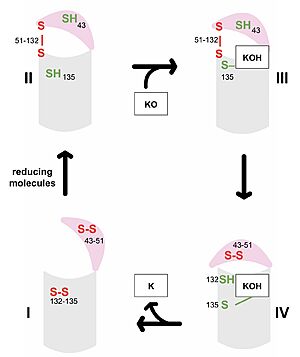
Figure 3. Catalytic Cycle of VKOR VKOR's luminal domain is represented by a the pink semicircle and the transmembrane domain is represented by the gray cylinder. Step I and II represent open conformations of VKOR and steps III and IV represent closed conformations.
The first step of the catalytic cycle (Figure 3) is the wild type open conformation, . This step is characterized by an open cap domain with disulfide bonds between cysteines 43 and 51 and between cysteines 132 and 135 [2]. The second step of the catalytic cycle is a partially oxidized open conformation, . This step is characterized by a disulfide bond between the luminal and transmembrane domain (Fig 3, step II). The transmembrane domain contains a free Cys135 and the luminal domain contains a free Cys43 [2]. Step II is labeled as open because no ligand exists within its binding pocket despite the disulfide bridge that connects the luminal and transmembrane domains. The next step of the cycle, , is also a closed structure with an intact disulfide bond between Cys51 and Cys132. Cys135 is not involved in a disulfide bridge and assists with substrate binding by forming a stable adduct with KOH or K. This binding induces the closed conformation and uses Cys43 in the luminal membrane for electron transfer [2]. of the catalytic cycle is the last closed conformation. The Cys51-Cys132 bond is broken as Cys43 bonds with Cys51, recreating the disulfide bridge pattern of the open state. Cys132 is then free to bond with Cys135, releasing the product that was bound to the Cys135. Overall the catalytic cycle of VKOR is dependent on open and closed conformational changes of the protein and ultimately is used to generate vitamin K from vitamin K epoxide [2].
Medical Relevance
Warfarin
Warfarin is the most widely prescribed oral anticoagulant and targets blood clotting via inhibition of vitamin K epoxide reductase. The FDA approved uses for cardiac conditions (myocardial infarction, atrial fibrillation) as well as for deep vein thrombosis and pulmonary embolism. Due to the inhibition of the normal blood clotting cycle, patients taking warfarin are at risk for hemorrhage which can occur anywhere in the body. [3]
Warfarin is a of Vitamin K and acts as a competitive inhibitor. There are around 30 known missense mutations that lead to warfarin resistance in patients, but these mutations do not affect Vitamin K binding for reasons which are not yet fully understood. Such patients require higher doses of warfarin to reach therapeutic level or require a different anticoagulant drug. [4]
Warfarin Dependence on Catalytic Cysteines
The anticoagulant warfarin works by inhibiting VKOR (See "Medical Relevance"). Warfarin binding also depends on the catalytic cysteines. Warfarin is able to bind to the fully oxidized open form of VKOR as shown in . Once Warfarin binds, VKOR is considered to be in a closed conformation since the substrate cannot enter, despite the lack of disulfide bridge changes. Warfarin can also bind to the partially oxidized form of VKOR as shown in .
Superwarfarins

Figure 4. Warfarin and Brodifacoum
More potent warfarin derivatives, called superwarfarins, are used as rodenticides. Superwarfarins have bulkier side chains that allow them to stay bound to VKOR for long periods of time, causing prolonged and uncontrolled bleeding. The duration of one superwarfarin, brodifacoum, has been reported as 15-30 days [5] vs. the clinical warfarin duration of 2-5 days[3].




